Taxonomía
GTDB-tk
GTDB-Tk es una herramienta que asigna taxonomía a genomas utilizando la base de datos GTDB (Genome Taxonomy Database). Basado en árboles filogenéticos y medidas de ANI (Average Nucleotide Identity), GTDB-Tk clasifica genomas bacterianos y arqueanos, proporciona una taxonomía coherente y actualizada. Se utiliza mucho en el análisis de genomas y metagenomas.
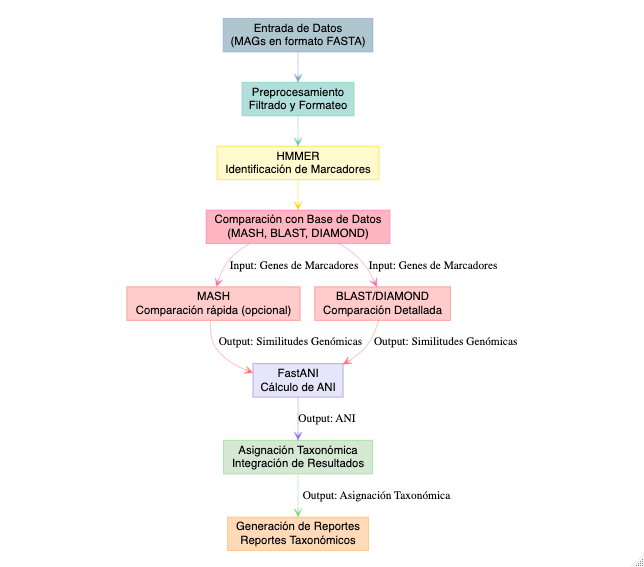
Recordemos que ya tenemos un set de bins refinados y desreplicados. Ahora vamos a asignarles identidad taxonómica, para ello vamos a correr GTDB-tk
Activa el ambiente
conda activate gtdbtk-2.1.1
El directorio de resultados para gtdbtk ya lo tienes en tu carpeta de resultados.
Para colocar los bins refinados y renombrados ejecuta el script src/copiar_renombrarbins.sh:
bash src/copiar_renombrarbins.sh
Ahora si, vamos a correr gtdbtk …
#pip install numpy==1.19.5
nohup gtdbtk classify_wf --genome_dir results/10.gtdbtk/bins/ --out_dir results/10.gtdbtk/ --cpus 6 -x fasta > outs/10.gtdbtk.nohup &
No olvides desactivar el ambiente
conda deactivate
Resultado de GTDB-Tk
Si gtdbtk está tomando mucho tiempo puedes parar el proceso con
ctrl + Cen tu teclado. El resultado final se encuentra en el directorio y archivo:results/10.gtdbtk/gtdbtk.bac120.summary.tsv.
Después de ejecutar GTDB-tk, continuaremos en R para visualizar los datos.
Crea un proyecto de R dentro del directorio results/10.gtdbtk/
library(tidyverse)
library(ggplot2)
# Leer la tabla ------------------------------------------------------------####
GTDBtk <- read.table("gtdbtk.bac120.summary.tsv",
sep = "\t", header = TRUE, na.strings = "",
stringsAsFactors = FALSE) %>%
as_tibble()
# Transformar datos --------------------------------------------------------####
pozol_gtdbtk <- GTDBtk %>%
select(user_genome, classification) %>%
separate(classification, c(
"Domain", "Phylum", "Class", "Order", "Family", "Genus", "Species"),
sep = ";") %>%
rename(Bin_name = user_genome) %>%
unite(Bin_name_2, c("Bin_name", "Phylum"), remove = FALSE) %>%
select(Bin_name, Domain, Phylum, Class, Order, Family, Genus, Species)
# Guardamos los datos en un archivo de metadatos ---------------------------####
write.table(pozol_gtdbtk, file = "Metadatos.txt",
sep = "\t", quote = FALSE, row.names = FALSE, col.names = TRUE)
# Visualización de Datos ---------------------------------------------------####
GTDBtk_barplot <- pozol_gtdbtk %>%
count(Phylum, Genus) %>%
rename(Number_of_MAGs = n) %>%
ggplot(aes(x = Phylum, y = Number_of_MAGs, fill = Genus)) +
geom_bar(stat = "identity", position = position_dodge()) +
theme_minimal()
GTDBtk_barplot
Discusión
Qué otra estrategias implementarías para la clasificación taxonómica?
Ejercicio 3
Ahora te toca a tí.
- Reúnanse en equipos y obtengan la clasificación taxonómica de los MAGs que obtuvieron con la muestra que les tocó.
- Discutan cada resultado obtenido.
- En la carpeta compartida de Drive busquen la diapositiva para el Ejercicio 3.
- En la diapositiva correspondiente resuman sus resultados obtenidos.
Tiempo de actividad (30 min)
Tiempo de presentación de resultados (2 min por equipo)
Metabolismo
PROKKA
Prokka es una herramienta útil, usa diferentes programas para predecir genes, secuencias codificantes, tRNAs, rRNAs. Hace la traducción de CDS a aminoácidos y asigna funciones usando varias bases de datos.
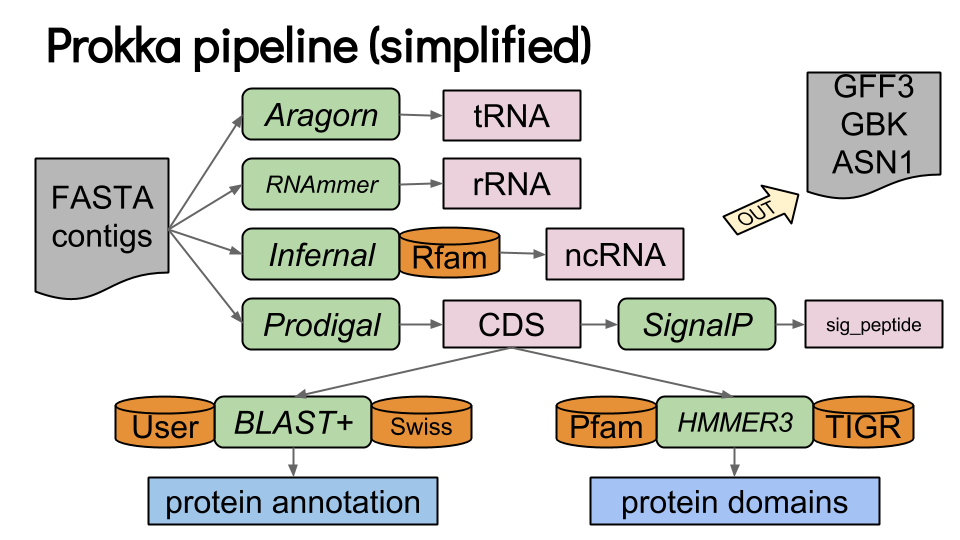
Para correrlo vamos a activar el ambiente en el que se aloja.
conda activate Prokka_Global
Tenemos el ambiente activo, ahora vamos a crear un directorio de resultados para prokka.
mkdir -p results/11.prokka
Para correrlo, podemos hacer un ciclo que nos permita anotar todos los bins. E
Escribe este ciclo en un script de bash que se llame 11.prokka.sh dentro del directorio de scripts src/.
#!/usr/bin/bash
# Ciclo for que ejecuta prokka en cada MAG refinado y desreplicado
for FASTA in $(ls results/10.gtdbtk/bins/); do
LOCUSTAG=$(basename $FASTA .fasta)
prokka --locustag "${LOCUSTAG}_Scaffold" --prefix $LOCUSTAG --addgenes \
--addmrna --cpus 4 --outdir "results/11.prokka/$LOCUSTAG" "results/10.gtdbtk/bins/$FASTA"
done
Y ahora córrelo asi:
nohup bash src/11.prokka.sh > outs/11.prokka.nohup &
Explora
Mientras prokka se ejecuta en los bins que obtuviste, despliega la ayuda y discute: ¿qué argumentos quitarías o agregarías?
Cuáles te llamaron la atención?
Responde:
¿Cuántos genes y cuántos CDS tiene el bin 48hBin01? Tip: usa
grep -c '>'¿En el contexto del pozol, cómo detectarías bins que estén degradando el maíz?
Desactivemos el ambiente:
conda deactivate
KofamScan
Ahora que tenemos las proteínas predichas vamos a obtener más anotaciones útiles, usaremos kofam para esto.
KofamScan es una herramienta de anotación, usa la base de datos KOfam de KEGG para obtener información sobre los genes que participan en diferentes rutas metabólicas.
Otras herramientas de anotación funcional
También puedes explorar:
- CAZy y su CAZYpedia.
- DRAM
- EggNOG-Mapper
Ejemplo de como correr KOfamScan
KofamScan requiere mucho tiempo de ejecución.
Para efectos del taller nosotros ya lo corrimos y te proporcionaremos los resultados.
Pero te dejamos el bloque de código que usamos para este paso:
mkdir -p results/12.kofam for FAA in $(ls results/11.prokka/*/*.faa); do name=$(basename $FAA .faa) exec_annotation $FAA \ -o results/12.kofam/"$name.txt" \ --report-unannotated \ --cpu 4 \ --tmp-dir results/12.kofam/"tmp$name" \ -p /home/alumno1/kofam/db/profiles/ \ -k /home/alumno1/kofam/db/ko_list done # remover los directorios temporales #rm -r results/12.kofam/tmp*
Estos resultados ya los tienes en el directorio results/12.kofam
Y ahora que ya tenemos los identificadores de KO para cada proteína, vamos a filtrar y graficar el metabolismo de los bins.
RbiMs
RbiMs es un paquete de R muy útil para obtener la anotación de cada KEGG ID y generar plots de esta información. Puede trabajar con anotaciones de KOFAM, Interpro, PFAM o CAZY.
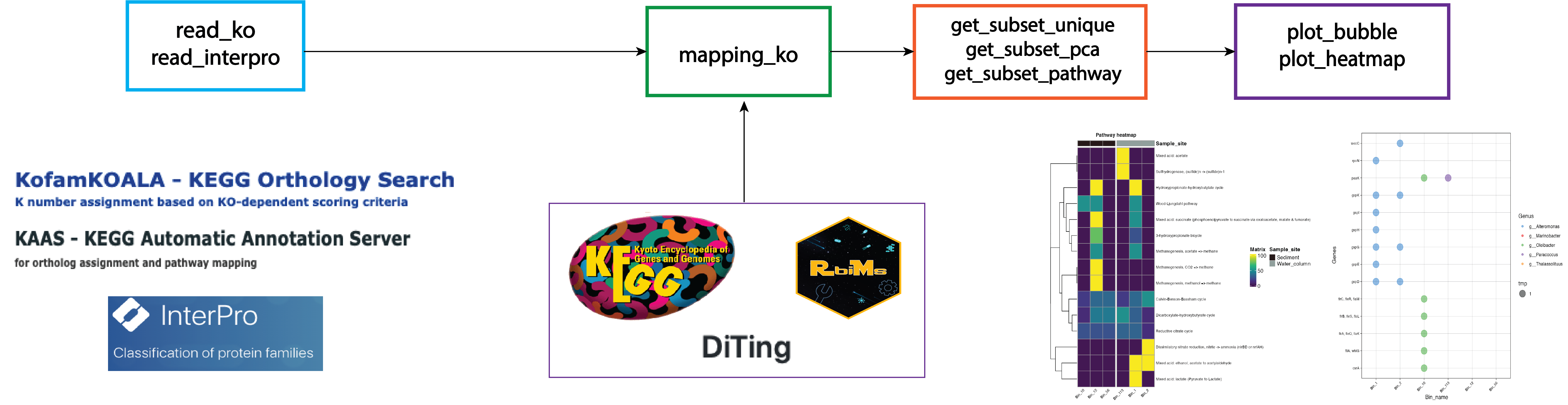
Vamos al editor de Rstudio para correr RbiMs ✨
library(tidyverse)
library(tidyr)
library(rbims)
library(readxl)
setwd("/home/alumnoX/taller_metagenomica_pozol/")
#A continuación, leemos los resultados de KEGG
pozol_table <- read_ko(data_kofam = "results/12.kofam/")
#y los mapeamos con la base de datos de KEGG:
pozol_mapp <- mapping_ko(pozol_table)
#Nos centraremos en las vías metabólicas relacionadas con la biosintesis de aminoacidos y vitaminas:
Overview <- c("Biosynthesis of amino acids", "Vitamin B6 metabolism")
Aminoacids_metabolism_pozol <- pozol_mapp %>%
drop_na(Module_description) %>%
get_subset_pathway(Pathway_description, Overview)
#Visualizamos los datos con un gráfico de burbujas:
plot_bubble(tibble_ko = Aminoacids_metabolism_pozol,
x_axis = Bin_name,
y_axis = Pathway_description,
analysis = "KEGG",
calc = "Percentage",
range_size = c(1, 10),
y_labs = FALSE,
x_labs = FALSE)
#Añadiremos metadatos, como la taxonomía:
Metadatos <- read_delim("results/10.gtdbtk/Metadatos.txt", delim = "\t")
#Y generaremos un gráfico de burbujas con metadatos:
plot_bubble(tibble_ko = Aminoacids_metabolism_pozol,
x_axis = Bin_name,
y_axis = Pathway_description,
analysis = "KEGG",
data_experiment = Metadatos,
calc = "Percentage",
color_character = Family,
range_size = c(1, 10),
y_labs = FALSE,
x_labs = FALSE)
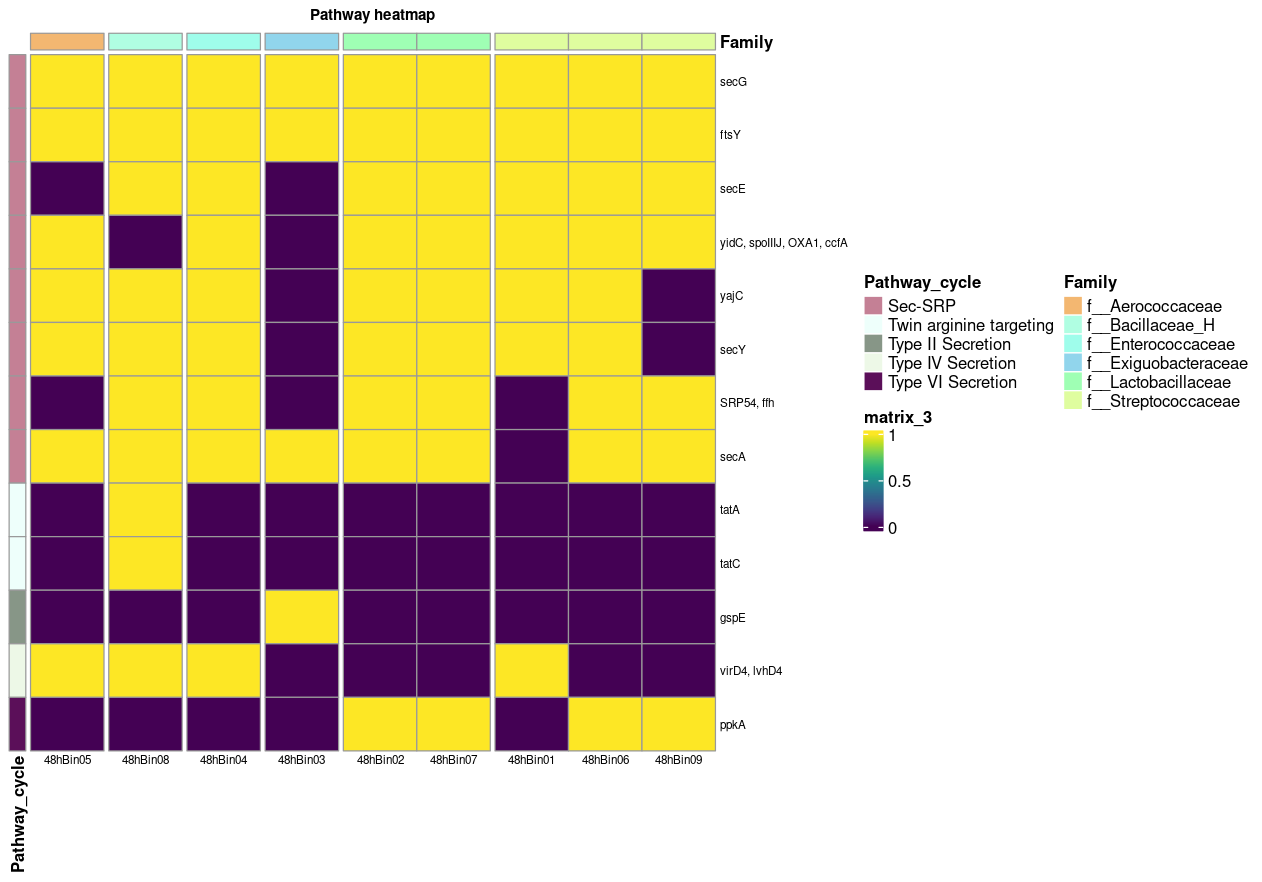
# Exploración de una Vía Específica
# podemos explorar una sola vía, como el “Secretion system,” y crear un mapa de calor para visualizar los genes relacionados con esta vía:
Secretion_system_pozol <- pozol_mapp %>%
drop_na(Cycle) %>%
get_subset_pathway(Cycle, "Secretion system")
#Y, finalmente, generamos un mapa de calor:
plot_heatmap(tibble_ko = Secretion_system_pozol,
y_axis = Genes,
analysis = "KEGG",
calc = "Binary")
#También podemos agregar metadatos para obtener una visión más completa:
plot_heatmap(tibble_ko = Secretion_system_pozol,
y_axis = Genes,
data_experiment = Metadatos,
order_x = Family,
analysis = "KEGG",
calc = "Binary")
plot_heatmap(tibble_ko = Secretion_system_pozol,
y_axis = Genes,
data_experiment = Metadatos,
order_y = Pathway_cycle,
order_x = Family,
analysis = "KEGG",
calc = "Binary")
# Explorar
colnames(pozol_mapp)
unique(pozol_mapp$Cycle)
Ejercicio 4
- Reúnanse en equipos y exploren la tabla de mapeo de RbiMs
- Seleccionen alguna via y obtenga un plot con lo que quieran incluir.
- En la carpeta compartida de Drive busquen la diapositiva para el Ejercicio 4.
- En la diapositiva correspondiente resuman sus resultados obtenidos.
Tiempo de actividad (15 min)
Tiempo de presentación de resultados (2 min por equipo)
Antismash
Adicionalmente podrías anotar el metabolismo secundario de los bins siguiendo el flujo de análisis propuestos en la lección de Minería Genómica de Software Carpentry.