Mapeo
En los días anteriores aprendieron a evaluar la calidad de las lecturas, filtrarlas y ensamblarlas, por lo que este apartado comenzará con un ensamble ya generado.
De acuerdo con el flujo de análisis (Figura 1), debemos partir de un ensamble, mapear las lecturas y obtener un archivo de profundidad de cada contig en el ensamble.
Archivos necesarios para hacer binning
El proceso de ensamble y mapeo son demandantes en tiempo de ejecución y recursos. Así que nos dimos a la tarea de generar el ensamble y el archivo de profundidad para comenzar directamente con el binning. El ensamble lo corrimos con Megahit, te dejamos la línea que usamos:
# Megahit megahit -1 data/full/48hrs_sm_R1.fastq -2 data/full/48hrs_sm_R2.fastq --min-contig-len 500 -t 40 --presets meta-sensitive --out-prefix 48hrs -o results/02.assembliesfull/megahit/48hrs # Con Metaspades podría ser asi: spades.py --meta -1 data/48hrs_sm_R1_5M.fastq -2 data/48hrs_sm_R2_5M.fastq -o results/02.assemblies/metaspades/48hrsEl mapeo lo corrimos con bowtie2 que es una herramienta confiable y muy utilizada para alinear lecturas cortas a una referencia, en nuestro caso, la referencia es el ensamble metagenómico de la muestra de 48hrs. Bowtie2 genera un archivo de mapeo (SAM) que debe convertirse a un formato binario (BAM), para esta conversión usamos samtools que contiene multiples subherramientas para trabajar con archivos de mapeos. Para generar este archivo se utilizaron las siguientes lineas de código:
# Formatear el ensamble bowtie2-build results/02.ensambles/megahit/48hrs/48hrs.fasta results/03.profundidad/48hrs --threads 40 # Mapear las lecturas contra el ensamble bowtie2 -x results/03.profundidad/48hrs -1 data/48hrs_sm_R1.fastq -2 data/48hrs_sm_R2.fastq -p 40 -S results/03.profundidad/48hrs.sam # Convertir de SAM a BAM y ordenar samtools view -Sb -O BAM -@ 40 results/03.profundidad/48hrs.sam | samtools sort -@ 40 -o results/03.profundidad/48hrs_sorted.bam # Obtener el índice samtools index results/03.profundidad/48hrs_sorted.bamYa que generamos el archivo bam ordenado y el índice, obtuvimos un archivo con la información de cobertura de cada contig dentro del ensamble, este archivo de profundidad se generó con
jgi_summarize_bam_contig_depthsque es una herramienta desarrollada por el JGI.# Obtener el archivo de profundidad de cada contig jgi_summarize_bam_contig_depths --outputDepth results/03.profundidad/48hrs.mgh_depth.txt results/03.profundidad/48hrs_sorted.bam🔊 🔊 No las ejecutes, sólo son un ejemplo para que las puedas usar con tus propios datos en el futuro.
Ejercicio 1. ¿Qué información requieren los programas de binning?
Antes de comenzar, reúnete con tu equipo y juntos:
- Revisen nuevamente el contenido de los directorios
02.ensamblesy03.profundidad.txt- En una diapositiva expliquen el
flujo teóricoque se siguió para obtener los archivos que están en esos directorios.Usa esta liga de drive para ir trabajando durante el taller.
Sólo un miembro de cada equipo escriba en la presentación
Binning
🧬 Ahora si, vamos a agrupar los contigs del metaensamble en bins …
Metabat2
Metabat2 es una herramienta que agrupa los contigs tomando la cobertura de cada contig y calcula su composición nucleotídica.
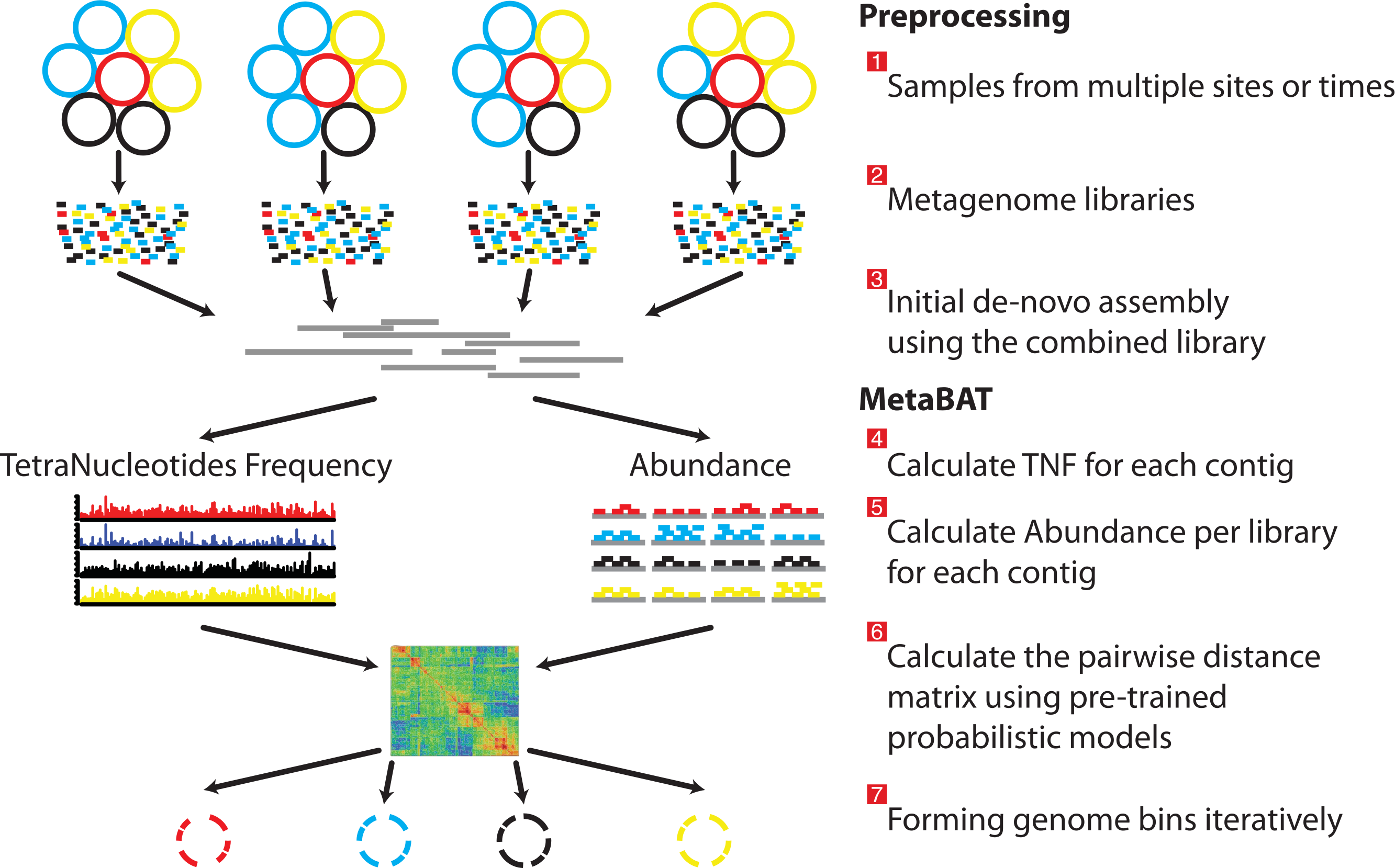
Metabat2. Kang et al., 2015. DOI:10.7717/peerj.1165
Para correr metabat necesitamos activar el ambiente conda donde se aloja.
conda activate binning
Ahora que ya tenemos el ambiente activado ejecutemos metabat:
metabat2 -i results/02.ensambles/48hrs.fasta -a results/03.profundidad/48hrs.mgh_depth.txt -o results/04.metabat/metabat -t 4 -m 1500
Se sabe que el valor mínimo de contig para reducir errores es 2000, lo puedes ver en la figura 6 de este artículo.
Responde
¿Cuántos bins se formaron? ¿Qué parámetros cambiarías o agregarías?
Solución
ls results/04.metabat/
metabat2 –-help
Ya que corrimos Metabat2 vamos a ejecutar MaxBin2, pero primero necesitamos desactivar el ambiente:
conda deactivate
MaxBin2
MaxBin2 agrupa los contigs de acuerdo a la información de cobertura, composición nucleotídica y marcadores de copia única.
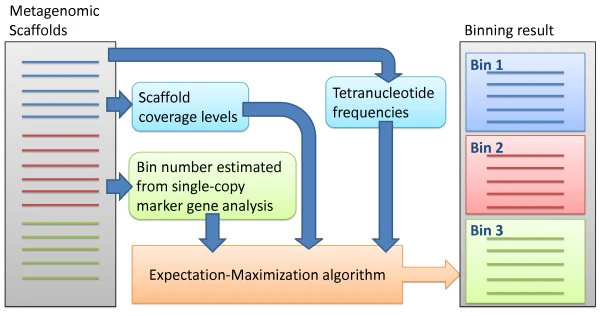
MaxBin2. Wu et al., 2014. https://doi.org/10.1186/2049-2618-2-26
Vamos a ejecutarlo, activemos el ambiente.
conda activate metagenomics
Crea el directorio para los resultados de MaxBin2
mkdir -p results/05.maxbin
Ahora si, vamos a ejecutarlo.
run_MaxBin.pl -thread 4 -min_contig_length 1500 -contig results/02.ensambles/48hrs.fasta -out results/05.maxbin/48hrs_maxbin -abund results/03.profundidad/48hrs.mgh_depth.txt
Responde
¿Cuántos bins se formaron? ¿Qué porcentaje de completitud tienen?
Solución
ls results/05.maxbin/*.fasta | wc -l
cat results/05.maxbin/48hrs_maxbin.summary | column -t
Desactiva el ambiente
conda deactivate
Vamb
VAMB utiliza una combinación de enfoques de aprendizaje profundo y técnicas de agrupamiento basándose en sus patrones de composición de nucleótidos y en la co-ocurrencia de sus frecuencias de cobertura.
Activa el ambiente binning
conda activate binning
Vamos a correr vamb, pero primero crea el directorio de resultados
mkdir -p results/06.vamb
Ejecutemos vamb:
nohup vamb --fasta results/02.ensambles/48hrs.fasta --jgi results/03.profundidad/48hrs.mgh_depth.txt --minfasta 500000 --outdir results/06.vamb/48hrs > outs/06.vamb.nohup &
Responde
Si quisieras recuperar los genomas de virus ¿Qué parámetro cambiarías?
Otros programas para binning
Recientemente se publicó COMEBin, que utiliza un enfoque distinto a lo que hemos usado en este tutorial. En el siguiente link encontrarás el manual y una explicación general sobre su funcionamiento.
Refinamiento
Ya corrimos tres programas de binning, pero, recordemos que los agrupamientos pueden tener errores:
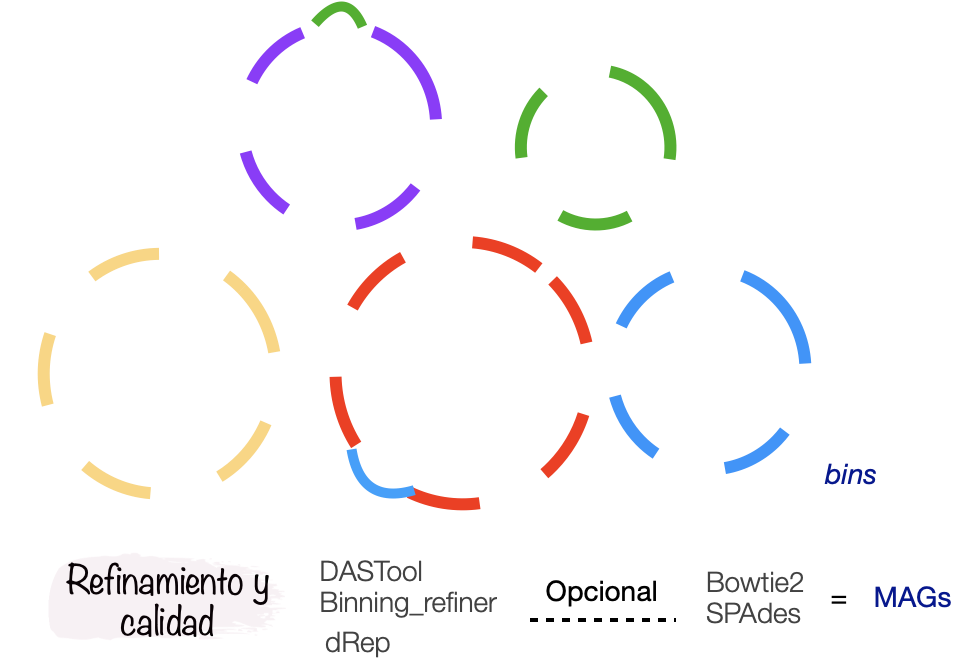
Para disminuir la contaminación e incrementar la completitud hay algunos programas que pueden ayudarnos. Entre ellos están Binning_refiner y DASTool.
CheckM
Antes de proceder al refinamiento es necesario tener claro cómo se evalúa la completitud y contaminación de los bins. Para esta evaluación se usa CheckM que se ha convertido en una herramienta estándar para la evaluación de la calidad de genomas y MAGs, y es usada por la mayoría de programas de refinamiento.
Para hacer esta evaluación, CheckM utiliza una serie de herramientas: tree organiza los genomas en un árbol de referencia. tree_qa evalúa la cantidad de genes marcadores filogenéticos y su ubicación en el árbol. El comando lineage_set crea un archivo de marcadores específicos de linaje, que se usa en el comando analyze para evaluar la integridad y contaminación de los genomas. Finalmente, el comando qa genera tablas que resumen la calidad de los genomas.
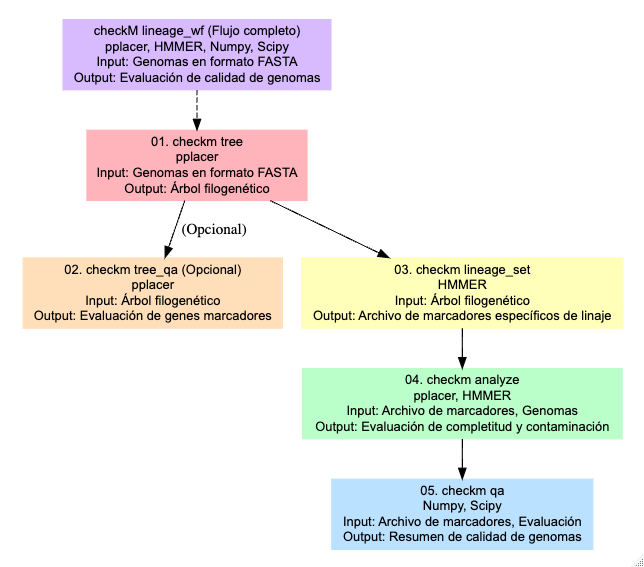
CheckM. Parks et al., 2015. https://genome.cshlp.org/content/25/7/1043.full
En este taller no vamos a correr CheckM porque los programas de refinamiento que usaremos ya lo corren de forma interna, sin embargo, es útil correrlo para tener una idea clara sobre la calidad de los bins que obtengamos.
Te dejamos la siguiente línea para que la uses en tus proyectos.
#ejemplo de como correrlo con los bins de vamb
#se debe activar el ambiente metagenomics
#checkm lineage_wf results/06.vamb/48hrs/bins results/checkm/ -x fna -t 4 -f results/checkm/checkm_vamb_bins.txt
Y una captura de ejemplo de como se ve la salida de CheckM:
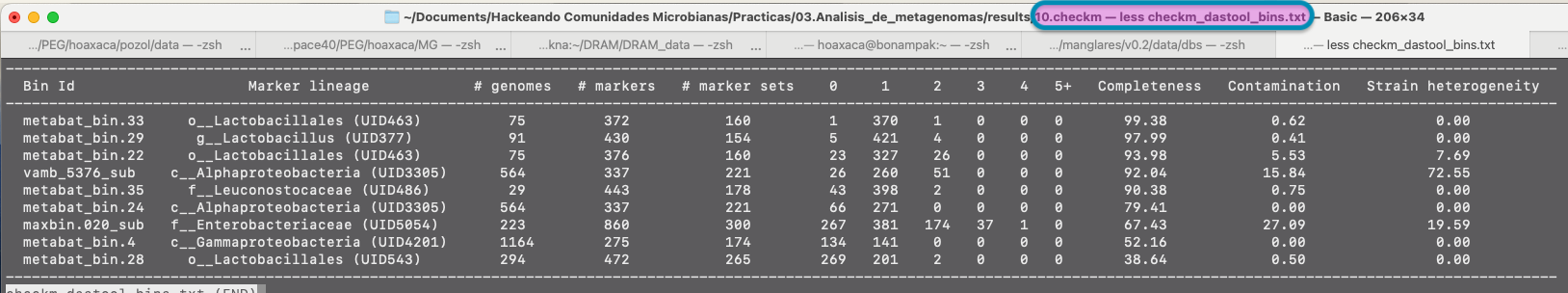
Y ahora si, a refinar los bins … 🥳
Binning_refiner
Binning_refiner se enfoca en refinar y fusionar los bins para mejorar la integridad y reducir la contaminación. Identifica bins que pueden representar el mismo genoma y los fusiona. Después elimina posibles contaminaciones, durante el proceso, Binning_refiner evalúa la calidad de los bins.
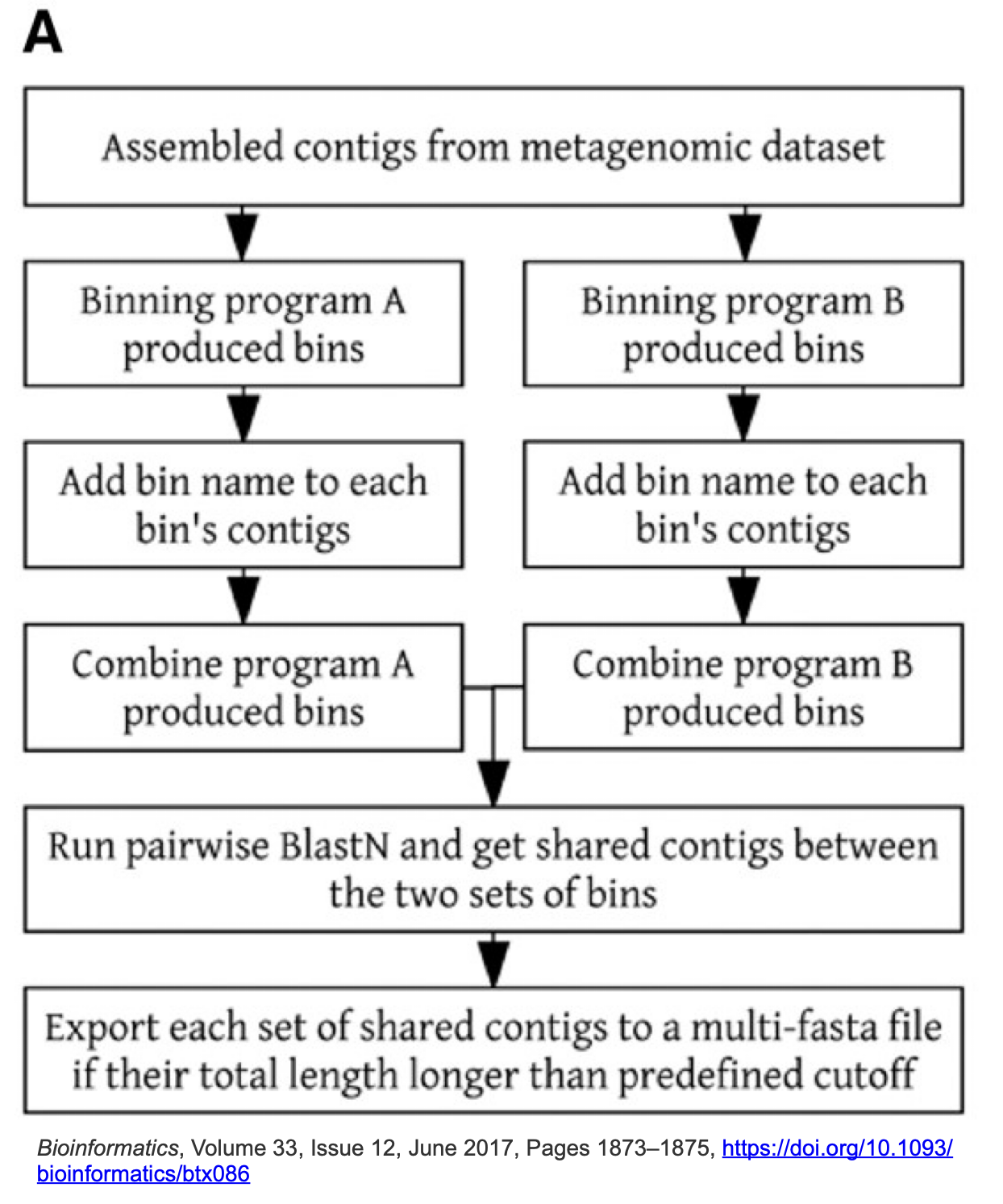
Binning_refiner. Wei-Zhi & Torsten, 2017. https://doi.org/10.1093/bioinformatics/btx086
Necesitamos crear el directorio de resultados para binning_refiner y un directorio con los bins generados por cada programa
mkdir -p results/07.binning_refiner/48hrsbins/{metabat,maxbin,vamb}
Ahora vamos a crear ligas simbólicas de los bins generados por cada herramienta.
#metabat
cd results/07.binning_refiner/48hrsbins/metabat/
ln -s ../../../04.metabat/*.fa .
#maxbin
cd ../maxbin/
ln -s ../../../05.maxbin/*.fasta .
# vamb
cd ../vamb/
ln -s ../../../06.vamb/48hrs/bins/*.fna .
#regresar
cd ../../
Ahora si, corramos Binning_refiner
Binning_refiner -i 48hrsbins/ -p 48hrs
Y regresemos a nuestro directorio principal
cd && cd taller_metagenomica_pozol/
Exploremos los resultados!
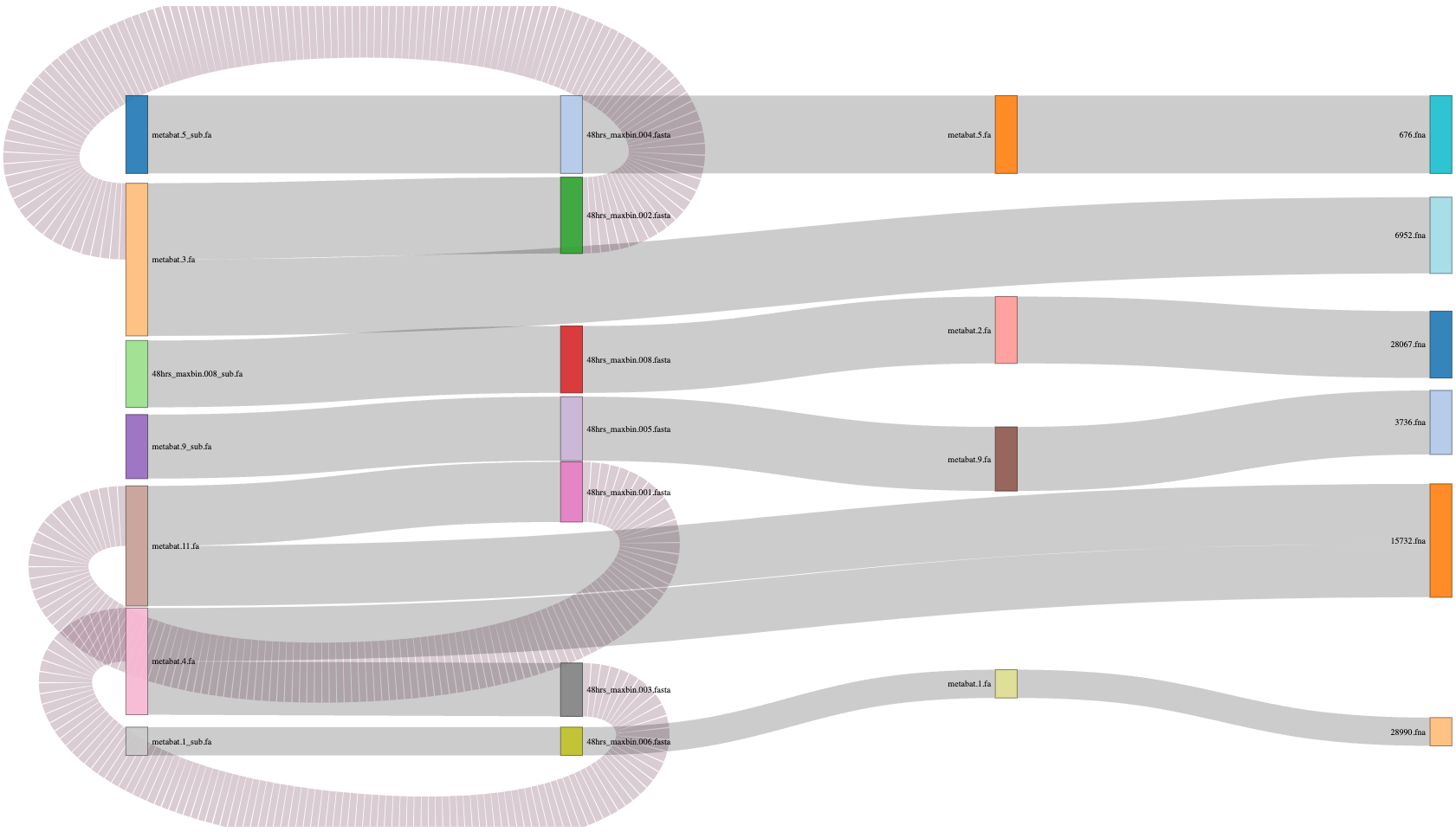
cat results/07.binning_refiner/48hrs_Binning_refiner_outputs/48hrs_sources_and_length.txt
Refined_bin Size(Kbp) Source
48hrs_1 1535.49 48hrs_maxbin.004.fasta,metabat.5.fa,676.fna
48hrs_2 1506.01 48hrs_maxbin.002.fasta,metabat.3.fa,6952.fna
48hrs_3 1319.12 48hrs_maxbin.008.fasta,metabat.2.fa,28067.fna
48hrs_4 1263.79 48hrs_maxbin.005.fasta,metabat.9.fa,3736.fna
48hrs_5 1185.25 48hrs_maxbin.001.fasta,metabat.11.fa,15732.fna
48hrs_6 1052.67 48hrs_maxbin.003.fasta,metabat.4.fa,15732.fna
48hrs_7 557.49 48hrs_maxbin.006.fasta,metabat.1.fa,28990.fna
Responde
¿Cuántos bins analizó Binning_refiner y cuántos quedaron después del refinamiento? ¿Hubo separación de algunos bins para formar otros?
Sankey plot
Puedes generar tu propio sankey plot para visualizar los resultados de Binning_refiner. Te dejamos el código por si te es útil.
código
# Cargar las librerias library(dplyr) library(networkD3) # revisa tu ubicación getwd() # OJO setwd("/home/ELALUMNOQUEERES/taller_metagenomica_pozol") # Cargar los datos sankey_data <- read.csv("results/07.binning_refiner/48hrs_Binning_refiner_outputs/48hrs_sankey.csv") # Crear una lista de nodos únicos nodes <- data.frame(name = unique(c(sankey_data$C1, sankey_data$C2))) # Crear el dataframe de enlaces links <- sankey_data %>% mutate(source = match(C1, nodes$name) - 1, target = match(C2, nodes$name) - 1, value = Length_Kbp) %>% select(source, target, value) # Crear el gráfico Sankey sankey_plot <- sankeyNetwork(Links = links, Nodes = nodes, Source = "source", Target = "target", Value = "value", NodeID = "name", fontSize = 12, nodeWidth = 30) # Mostrar el gráfico sankey_plot # Guardar library(htmlwidgets) saveWidget(sankey_plot, file = "48hrs_sankey_plot.html")
DASTool
DASTool es una herramienta utilizada para mejorar la calidad de los bins. Evalúa la integridad, combina los resultados de diferentes bineadores y por consenso selecciona los mejores bins de cada herramienta. Una vez que DASTool ha seleccionado los mejores bins, realiza un proceso de refinamiento para optimizar los resultados.
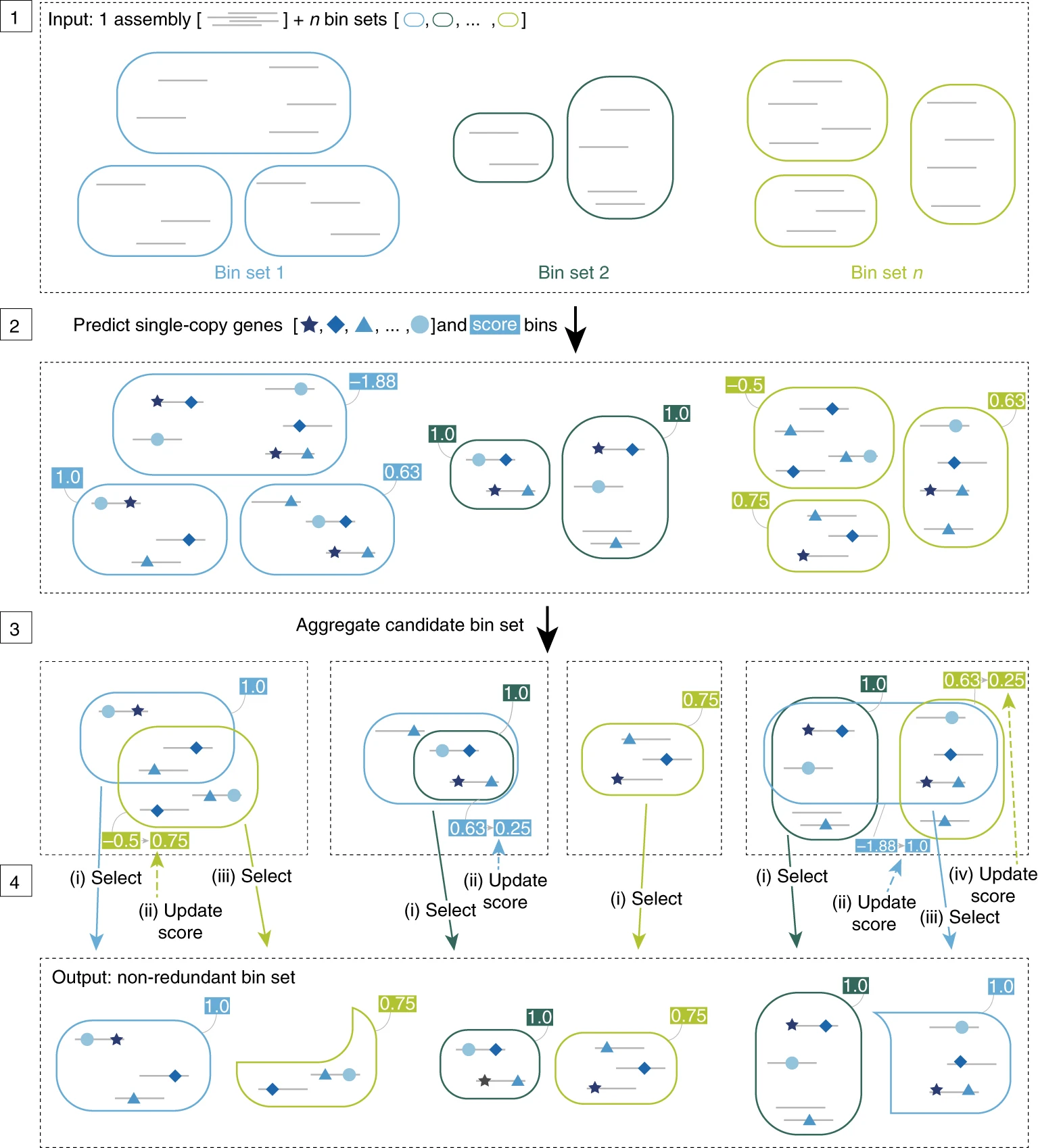
DASTool. Sieber et al., 2018. https://doi.org/10.1038/s41564-018-0171-1.
Vamos a correr DASTool …
Primero crea el directorio para los resultados
mkdir -p results/08.dastool
DASTool necesita como entrada un archivo tabular con información de los resultados de cada programa de binning.
Fasta_to_Contig2Bin.sh -i results/04.metabat/ -e fa > results/08.dastool/48hrs_metabat.dastool.tsv
Fasta_to_Contig2Bin.sh -i results/05.maxbin/ -e fasta > results/08.dastool/48hrs_maxbin.dastool.tsv
Fasta_to_Contig2Bin.sh -i results/06.vamb/48hrs/bins/ -e fna > results/08.dastool/48hrs_vamb.dastool.tsv
Ya que tenemos los archivos tsv podemos empezar con el refinamiento!! 🥳
DAS_Tool -i results/08.dastool/48hrs_metabat.dastool.tsv,results/08.dastool/48hrs_maxbin.dastool.tsv,results/08.dastool/48hrs_vamb.dastool.tsv -l metabat,maxbin,vamb -c results/02.ensambles/48hrs.fasta -o results/08.dastool/48hrs -t 4 --write_bins
Responde
¿Cuántos bins quedaron después del refinamiento con DASTool? ¿Más o menos que con Binning_refiner? ¿Qué calidad tienen?
Desreplicación
dRep
La desreplicación es el proceso de identificar conjuntos de genomas que son “iguales” en una lista de genomas y eliminar todos los genomas excepto el “mejor” de cada conjunto redundante. dRep es una herramienta útil para esto. Utiliza distancias Mash y ANIm para discriminar entre MAGs iguales.
DASTool. Olm et al., 2017. https://academic.oup.com/ismej/article/11/12/2864/7537826.
Ya que tenemos los resultados de los dos refinadores ejecutaremos dRep para desreplicar y seleccionar el mejor representante de cada bin.
Primero vamos a crear el directorio de resultados para dRep.
mkdir -p results/09.drep/bins
Y entraremos al directorio bins dentro del directorio de resultados para colocar los bins que queremos comparar. En este caso los generados por ambos refinadores (pero podrían ser los bins refinados de cada punto de muestreo).
cd results/09.drep/bins/
Con las siguientes lineas podemos copiar los bins en este directorio:
Primero los de DASTool
for i in $(ls ../../08.dastool/48hrs_DASTool_bins/*.fa); do
name=$(basename $i .fa); cp $i $name".fasta"
done
Y ahora los de Binning_refiner
cp ../../07.binning_refiner/48hrs_Binning_refiner_outputs/48hrs_refined_bins/*.fasta .
Ya que los copiamos, regresemos al directorio principal.
cd && cd taller_metagenomica_pozol/
Y ahora si, vamos a correr dRep …
export PATH=/miniconda3/envs/metagenomics/bin:$PATH
nohup dRep dereplicate results/09.drep/ -d -comp 50 -con 10 --SkipSecondary -g results/09.drep/bins/*.fasta > outs/09.drep.nohup &
Nota
El argumento
--SkipSecondaryno se aconseja poner, en la vida real queremos que se hagan todas las agrupaciones para discriminar genomas. En este ejemplo fue necesario ponerlo porque no logramos llamar a ANIm
Este es uno de los plots generados por dRep, que representa los mejores bins desreplicados.
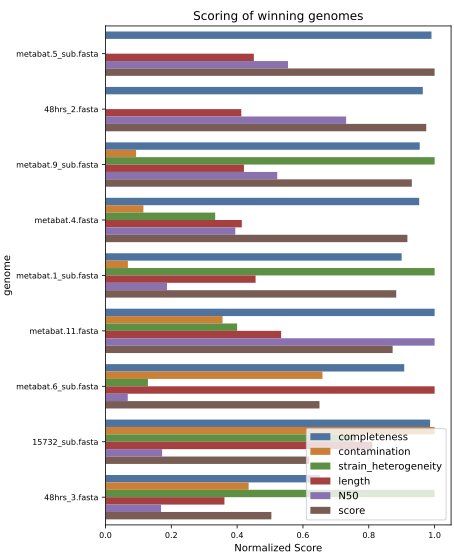
Vamos a desactivar el ambiente de dRep
conda deactivate
Responde
¿Cuántos clústers se formaron? ¿Cuántos MAGs son únicos?
🧠 Discusión
Para tomar en cuenta
- En la vida real, si el proyecto de metagenómica que estás desarrollando tiene librerías de diferentes muestras usarías dRep entre todos los conjuntos de bins ya refinados para no tener redundancia de genomas.
- Qué harías si antes de desreplicar tienes un bin que tiene 98 % de completitud y 11 % de contaminación?. dRep en automático lo descartaría.
Propondrías alguna manera para quedarte con este bin y curarlo para reducir su contaminación?
Por suerte hay más programas que pueden ayudarnos a curar nuestros bins manualmente, una herramienta útil para esto es mmgenome2
Ya que tenemos los bins refinados y desreplicados opcionalmente podrías reensamblarlos. La manera sería mapear las lecturas de toda la muestra a los bins finales y con las lecturas mapeadas y el bin, generar un ensamble genómico para cada uno. Con esta aproximación se genera un MAG más pulido y la contaminación se reduce.
Aunque en muchos reportes verás que los autores reensamblan sus MAGs, en otros no lo hacen y no hacerlo no está mal, pero hacerlo mejora la calidad.
🧬🦠🥳 Ahora te toca a tí
Ejercicio 2
Ahora te toca a tí.
- Reúnanse en equipos y repliquen todo el flujo hasta este punto con la muestra que les toca.
- Discutan cada resultado obtenido.
- En la carpeta compartida de Drive busquen la diapositiva para el Ejercicio 2.
- En la diapositiva correspondiente resuman sus resultados obtenidos.
Tiempo de actividad (2 hr)
Tiempo de presentación de resultados (5 min por equipo)