Exploring Taxonomy with R
Overview
Teaching: 20 min
Exercises: 5 minQuestions
How can I use my taxonomic assignment results to analyze?
Objectives
Comprehend which libraries are required for analysis of the taxonomy of metagenomes.
Create and manage a Phyloseq object.
Creating lineage and rank tables
In this episode, we will use RStudio to analyze our microbial samples. You do not have to install anything, you already have an instance on the cloud ready to be used.
Packages like Qiime2, MEGAN, Vegan, or Phyloseq in R allow us to analyze diversity and abundance by
manipulating taxonomic assignment data. In this lesson, we will use Phyloseq. In order to do so, we need to generate
an abundance matrix from the Kraken output files. One program widely used for this purpose is kraken-biom.
To do this, we could go to our now familiar Bash terminal, but RStudio has an integrated terminal that uses the same language as the one we learned in the Command-line lessons, so let us take advantage of it. Let us open RStudio and go to the Terminal tab in the bottom left panel.
Kraken-biom
Kraken-biom is a program that creates BIOM tables from the Kraken output.
In order to run Kraken-biom, we have to move to the folder where our taxonomic data files are located:
$ cd ~/dc_workshop/taxonomy
First, we will visualize the content of our directory by the ls command.
$ ls
JC1A.kraken JC1A.report JP41.report JP4D.kraken JP4D.report mags_taxonomy
The kraken-biom program is installed inside our metagenomics environment, so let us activate it.
$ conda activate metagenomics
Let us take a look at the different flags that kraken-biom has:
$ kraken-biom -h
usage: kraken-biom [-h] [--max {D,P,C,O,F,G,S}] [--min {D,P,C,O,F,G,S}]
[-o OUTPUT_FP] [--otu_fp OTU_FP] [--fmt {hdf5,json,tsv}]
[--gzip] [--version] [-v]
kraken_reports [kraken_reports ...]
Create BIOM-format tables (http://biom-format.org) from Kraken output
(http://ccb.jhu.edu/software/kraken/).
.
.
.
By a close look at the first output lines, it is noticeable that we need a specific output
from Kraken: the .reports.
With the following command, we will create a table in Biom format called cuatroc.biom. We will include the two samples we have been working with (JC1A and JP4D) and a third one (JP41) to be able to perform specific analyses later on.
$ kraken-biom JC1A.report JP4D.report JP41.report --fmt json -o cuatroc.biom
If we inspect our folder, we will see that the cuatroc.biom file has been created. This biom object contains both the abundance and the ID (a number) of each OTU.
With this result, we are ready to return to RStudio’s console and begin to manipulate our
taxonomic-data.
Command line prompts
Note that you can distinguish the Bash terminal from the R console by looking at the prompt. In Bash is the
$sign, and in R is the>sign.
Creating and manipulating Phyloseq objects
Load required packages
Phyloseq is a library with tools to analyze and plot your metagenomics samples’ taxonomic assignment and abundance information. Let us install phyloseq (This instruction might not work on specific versions of R) and other libraries required for its execution:
> if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
> BiocManager::install("phyloseq") # Install phyloseq
> install.packages(c("RColorBrewer", "patchwork")) #install patchwork to chart publication-quality plots and readr to read rectangular datasets.
Once the libraries are installed, we must make them available for this R session. Now load the libraries (a process needed every time we begin a new work session in R):
> library("phyloseq")
> library("ggplot2")
> library("RColorBrewer")
> library("patchwork")
Creating the phyloseq object
First, we tell R in which directory we are working.
> setwd("~/dc_workshop/taxonomy/")
Let us proceed to create the phyloseq object with the import_biom command:
> merged_metagenomes <- import_biom("cuatroc.biom")
Now, we can inspect the result by asking the class of the object created and doing a close inspection of some of its content:
> class(merged_metagenomes)
[1] "phyloseq"
attr("package")
[1] "phyloseq"
The “class” command indicates that we already have our phyloseq object.
Exploring the taxonomic labels
Let us try to access the data that is stored inside our merged_metagenomes object. Since a phyloseq object
is a special object in R, we need to use the operator @ to explore the subsections of data inside merged_metagenomes.
If we type merged_metagenomes@, five options are displayed; tax_table and otu_table are the ones
we will use. After writing merged_metagenomes@otu_table or merged_metagenomes@tax_table, an option of .Data
will be the one chosen in both cases. Let us see what is inside our tax_table:
> View(merged_metagenomes@tax_table@.Data)
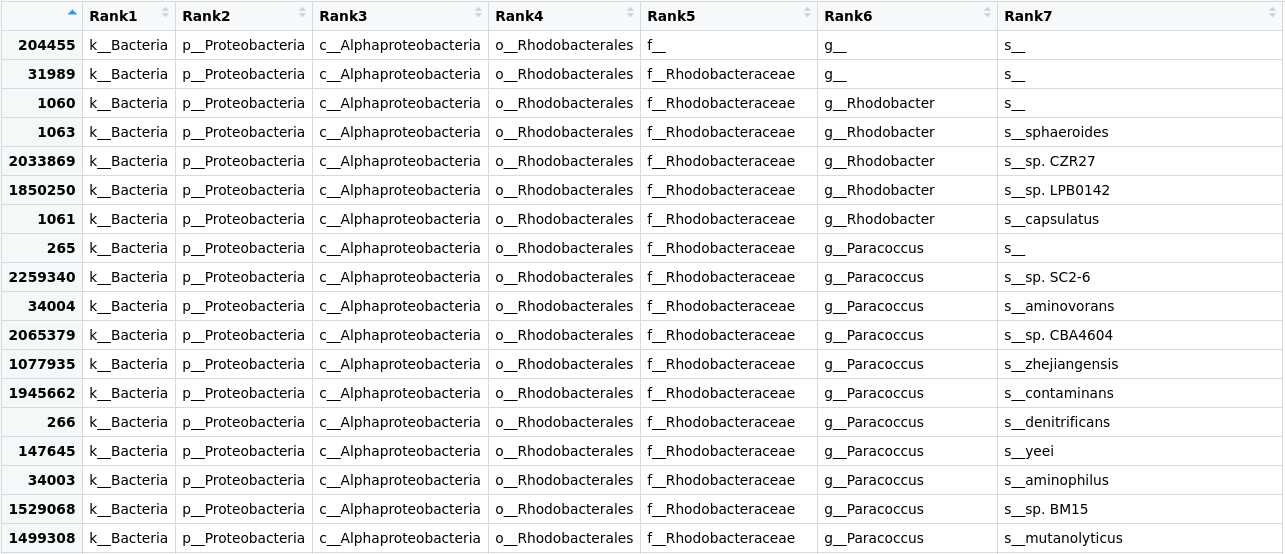 Figure 1. Table of the taxonomic labels from our
Figure 1. Table of the taxonomic labels from our merged_metagenomes object.
Here we can see that the tax_table inside our phyloseq object stores all the taxonomic labels corresponding to each OTU. Numbers in the row names of the table identify OTUs.
Next, let us get rid of some of the unnecessary characters in the OTUs id and put names to the taxonomic ranks:
To remove unnecessary characters in .Data (matrix), we will use the command substring(). This command helps extract or replace characters in a vector. To use the command, we have to indicate the vector (x) followed by the first element to replace or extract (first) and the last element to be replaced (last). For instance: substring (x, first, last). substring() is a “flexible” command, especially to select
characters of different lengths, as in our case. Therefore, it is not necessary to indicate “last”, so it will take the last position of the character by
default. Since a matrix is an arrangement of vectors, we can use this command. Each character in .Data is preceded by three spaces occupied by a
letter and two underscores, for example: o__Rhodobacterales. In this case, “Rodobacterales” starts at position 4 with an R. So, to remove the unnecessary characters, we will use the following code:
> merged_metagenomes@tax_table@.Data <- substring(merged_metagenomes@tax_table@.Data, 4)
> colnames(merged_metagenomes@tax_table@.Data)<- c("Kingdom", "Phylum", "Class", "Order", "Family", "Genus", "Species")
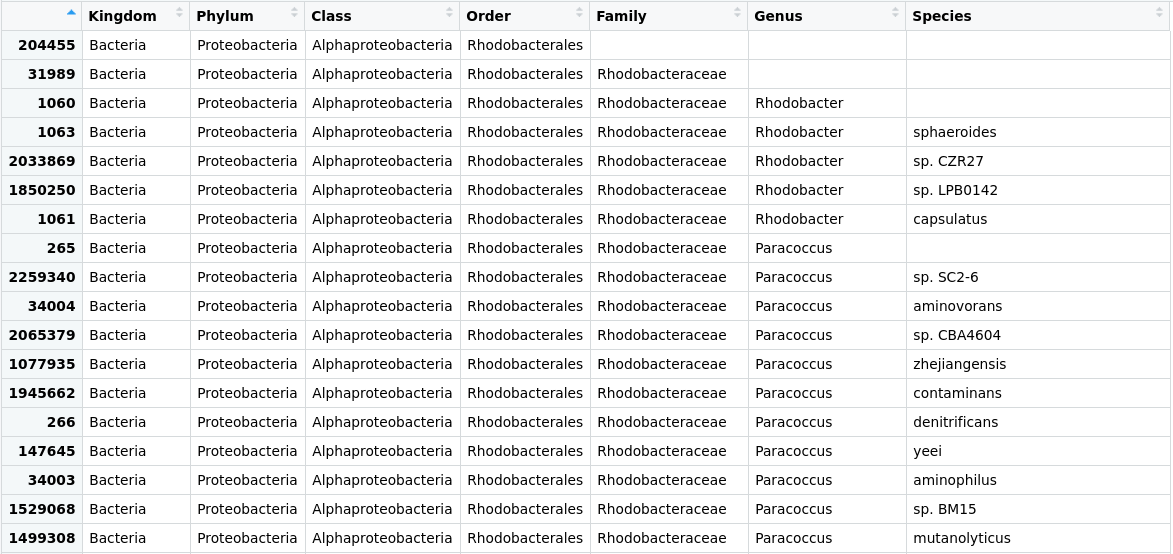 Figure 2. Table of the taxonomic labels from our
Figure 2. Table of the taxonomic labels from our merged_metagenomes object with corrections.
We will use a command named unique() to explore how many phyla we have. Let us see the result
we obtain from the following code:
> unique(merged_metagenomes@tax_table@.Data[,"Phylum"])
[1] "Proteobacteria" "Actinobacteria" "Firmicutes"
[4] "Cyanobacteria" "Deinococcus-Thermus" "Chloroflexi"
[7] "Armatimonadetes" "Bacteroidetes" "Chlorobi"
[10] "Gemmatimonadetes" "Planctomycetes" "Verrucomicrobia"
[13] "Lentisphaerae" "Kiritimatiellaeota" "Chlamydiae"
[16] "Acidobacteria" "Spirochaetes" "Synergistetes"
[19] "Nitrospirae" "Tenericutes" "Coprothermobacterota"
[22] "Ignavibacteriae" "Candidatus Cloacimonetes" "Fibrobacteres"
[25] "Fusobacteria" "Thermotogae" "Aquificae"
[28] "Thermodesulfobacteria" "Deferribacteres" "Chrysiogenetes"
[31] "Calditrichaeota" "Elusimicrobia" "Caldiserica"
[34] "Candidatus Saccharibacteria" "Dictyoglomi"
Knowing phyla is helpful, but what we need to know is how many of our OTUs have been assigned to the phylum
Firmicutes?. Let´s use the command sum() to ask R:
> sum(merged_metagenomes@tax_table@.Data[,"Phylum"] == "Firmicutes")
[1] 580
Now, to know for that phylum in particular which taxa there are in a certain rank, we can also ask it to phyloseq.
> unique(merged_metagenomes@tax_table@.Data[merged_metagenomes@tax_table@.Data[,"Phylum"] == "Firmicutes", "Class"])
[1] "Bacilli" "Clostridia" "Negativicutes" "Limnochordia" "Erysipelotrichia" "Tissierellia"
Exploring the abundance table
Until now, we have looked at the part of the phyloseq object that stores the information about the taxonomy (at all the possible levels) of each OTU found in our samples. However, there is also a part of the phyloseq object that stores the information about how many sequenced reads corresponding to a certain OTU are in each sample. This table is the otu_table.
> View(merged_metagenomes@otu_table@.Data)
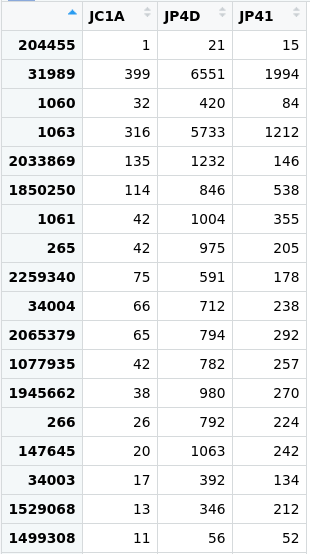 Figure 3. Table of the abundance of reads in the
Figure 3. Table of the abundance of reads in the merged_metagenomes object.
We will take advantage of this information later on in our analyses.
Phyloseq objects
Finally, we can review our object and see that all datasets (i.e., JC1A, JP4D, and JP41) are in the object. If you look at our Phyloseq object, you will see that there are more data types that we can use to build our object(
?phyloseq()), such as a phylogenetic tree and metadata concerning our samples. These are optional, so we will use our basic phyloseq object, composed of the abundances of specific OTUs and the names of those OTUs.
Exercise 1: Explore a phylum
Go into groups and choose one phylum that is interesting for your group, and use the learned code to find out how many OTUs have been assigned to your chosen phylum and what are the unique names of the genera inside it. がんばって! (ganbatte; good luck):
Solution
Change the name of a new phylum wherever needed and the name of the rank we are asking for to get the result. As an example, here is the solution for Proteobacteria:
sum(merged_metagenomes@tax_table@.Data[,"Phylum"] == "Proteobacteria")unique(merged_metagenomes@tax_table@.Data[merged_metagenomes@tax_table@.Data[,"Phylum"] == "Proteobacteria", "Genus"])
Exercise 2: Searching for the read counts
Using the information from both the
tax_tableand theotu_table, find how many reads there are for any species of your interest (one that can be found in thetax_table).
Hint: Remember that you can access the contents of a data frame with the["row_name", "column_name"]syntax.
がんばって! (ganbatte; good luck):Solution
Go to the
tax_table:> View(merged_metagenomes@tax_table@.Data)Take note of the OTU number for some species:
Figure 4. The row of the
tax_tablecorresponds to the species Paracoccus zhejiangensis.Search for the row of the
otu_tablewith the row name you chose.> merged_metagenomes@otu_table@.Data["1077935",]JC1A JP4D JP41 42 782 257
Key Points
kraken-biom formats Kraken output-files of several samples into the single
.biomfile that will be phyloseq input.The library
phyloseqmanages metagenomics objects and computes analyses.A phyloseq object stores a table with the taxonomic information of each OTU and a table with the abundance of each OTU.