Assessing Read Quality
Overview
Teaching: 30 min
Exercises: 20 minQuestions
How can I describe the quality of my data?
Objectives
Explain how a FASTQ file encodes per-base quality scores.
Interpret a FastQC plot summarizing per-base quality across all reads.
Use
forloops to automate operations on multiple files.
Bioinformatic workflows
When working with high-throughput sequencing data, the raw reads you get off the sequencer must pass through several different tools to generate your final desired output. The execution of this set of tools in a specified order is commonly referred to as a workflow or a pipeline.
An example of the workflow we will be using for our analysis is provided below, with a brief description of each step.
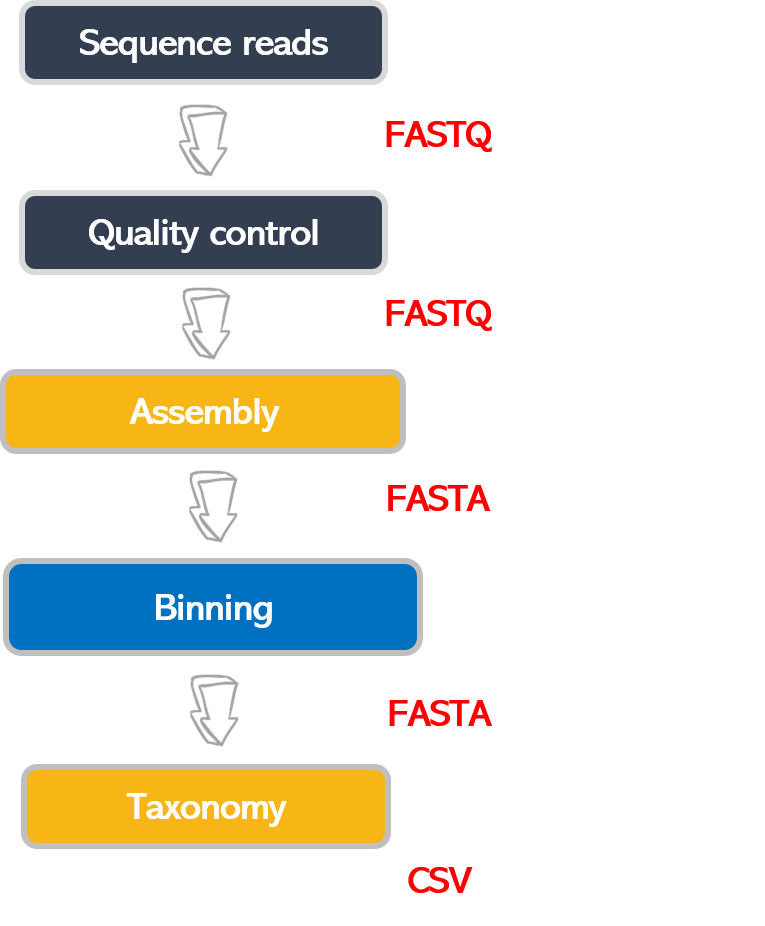
- Quality control - Assessing quality using FastQC and Trimming and/or filtering reads (if necessary)
- Assembly of metagenome
- Binning
- Taxonomic assignation
These workflows in bioinformatics adopt a plug-and-play approach in that the output of one tool can be easily
used as input to another tool without any extensive configuration. Having standards for data formats is what
makes this feasible. Standards ensure that data is stored in a way that is generally accepted and agreed upon
within the community. Therefore, the tools used to analyze data at different workflow stages are
built, assuming that the data will be provided in a specific format.
Quality control
We will now assess the quality of the sequence reads contained in our FASTQ files.
Details on the FASTQ format
Although it looks complicated (and it is), we can understand the FASTQ format with a little decoding. Some rules about the format include the following:
| Line | Description |
|---|---|
| 1 | Always begins with ‘@’ followed by the information about the read |
| 2 | The actual DNA sequence |
| 3 | Always begins with a ‘+’ and sometimes contains the same info as in line 1 |
| 4 | Has a string of characters which represent the quality scores; must have same number of characters as line 2 |
We can view the first complete read in one of the files from our dataset using head to look at
the first four lines. But we have to decompress one of the files first.
$ cd ~/dc_workshop/data/untrimmed_fastq/
$ gunzip JP4D_R1.fastq.gz
$ head -n 4 JP4D_R1.fastq
@MISEQ-LAB244-W7:156:000000000-A80CV:1:1101:12622:2006 1:N:0:CTCAGA
CCCGTTCCTCGGGCGTGCAGTCGGGCTTGCGGTCTGCCATGTCGTGTTCGGCGTCGGTGGTGCCGATCAGGGTGAAATCCGTCTCGTAGGGGATCGCGAAGATGATCCGCCCGTCCGTGCCCTGAAAGAAATAGCACTTGTCAGATCGGAAGAGCACACGTCTGAACTCCAGTCACCTCAGAATCTCGTATGCCGTCTTCTGCTTGAAAAAAAAAAAAGCAAACCTCTCACTCCCTCTACTCTACTCCCTT
+
A>>1AFC>DD111A0E0001BGEC0AEGCCGEGGFHGHHGHGHHGGHHHGGGGGGGGGGGGGHHGEGGGHHHHGHHGHHHGGHHHHGGGGGGGGGGGGGGGGHHHHHHHGGGGGGGGHGGHHHHHHHHGFHHFFGHHHHHGGGGGGGGGGGGGGGGGGGGGGGGGGGGFFFFFFFFFFFFFFFFFFFFFBFFFF@F@FFFFFFFFFFBBFF?@;@####################################
Line 4 shows the quality of each nucleotide in the read. Quality is interpreted as the probability of an incorrect base call (e.g., 1 in 10) or, equivalently, the base call accuracy (e.g., 90%). Each nucleotide’s numerical score’s value is converted into a character code where every single character represents a quality score for an individual nucleotide. This conversion allows the alignment of each individual nucleotide with its quality score. For example, in the line above, the quality score line is:
A>>1AFC>DD111A0E0001BGEC0AEGCCGEGGFHGHHGHGHHGGHHHGGGGGGGGGGGGGHHGEGGGHHHHGHHGHHHGGHHHHGGGGGGGGGGGGGGGGHHHHHHHGGGGGGGGHGGHHHHHHHHGFHHFFGHHHHHGGGGGGGGGGGGGGGGGGGGGGGGGGGGFFFFFFFFFFFFFFFFFFFFFBFFFF@F@FFFFFFFFFFBBFF?@;@####################################
The numerical value assigned to each character depends on the sequencing platform that generated the reads. The sequencing machine used to generate our data uses the standard Sanger quality PHRED score encoding, using Illumina version 1.8 onwards. Each character is assigned a quality score between 0 and 41, as shown in the chart below.
Quality encoding: !"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJ
| | | | |
Quality score: 01........11........21........31........41
Each quality score represents the probability that the corresponding nucleotide call is incorrect. These probability values are the results of the base calling algorithm and depend on how much signal was captured for the base incorporation. This quality score is logarithmically based, so a quality score of 10 reflects a base call accuracy of 90%, but a quality score of 20 reflects a base call accuracy of 99%. In this link you can find more information about quality scores.
Looking back at our read:
@MISEQ-LAB244-W7:156:000000000-A80CV:1:1101:12622:2006 1:N:0:CTCAGA
CCCGTTCCTCGGGCGTGCAGTCGGGCTTGCGGTCTGCCATGTCGTGTTCGGCGTCGGTGGTGCCGATCAGGGTGAAATCCGTCTCGTAGGGGATCGCGAAGATGATCCGCCCGTCCGTGCCCTGAAAGAAATAGCACTTGTCAGATCGGAAGAGCACACGTCTGAACTCCAGTCACCTCAGAATCTCGTATGCCGTCTTCTGCTTGAAAAAAAAAAAAGCAAACCTCTCACTCCCTCTACTCTACTCCCTT
+
A>>1AFC>DD111A0E0001BGEC0AEGCCGEGGFHGHHGHGHHGGHHHGGGGGGGGGGGGGHHGEGGGHHHHGHHGHHHGGHHHHGGGGGGGGGGGGGGGGHHHHHHHGGGGGGGGHGGHHHHHHHHGFHHFFGHHHHHGGGGGGGGGGGGGGGGGGGGGGGGGGGGFFFFFFFFFFFFFFFFFFFFFBFFFF@F@FFFFFFFFFFBBFF?@;@####################################
We can now see that there is a range of quality scores but that the end of the sequence is
very poor (# = a quality score of 2).
Exercise 1: Looking at specific reads
In the terminal, how would you show the ID and quality of the last read
JP4D_R1.fastq?
a)tail JP4D_R1.fastq
b)head -n 4 JP4D_R1.fastq
c)more JP4D_R1.fastq
d)tail -n4 JP4D_R1.fastq
e)tail -n4 JP4D_R1.fastq | head -n2Do you trust the sequence in this read?
Solution
a) It shows the ID and quality of the last read but also unnecessary lines from previous reads. b) No. It shows the first read's info. c) It shows the text of the entire file. d) This option is the best answer as it only shows the last read's information. e) It does show the ID of the last read but not the quality.@MISEQ-LAB244-W7:156:000000000-A80CV:1:2114:17866:28868 1:N:0:CTCAGA CCCGTTCTCCACCTCGGCGCGCGCCAGCTGCGGCTCGTCCTTCCACAGGAACTTCCACGTCGCCGTCAGCCGCGACACGTTCTCCCCCCTCGCATGCTCGTCCTGTCTCTCGTGCTTGGCCGACGCCTGCGCCTCGCACTGCGCCCGCTCGGTGTCGTTCATGTTGATCTTCACCGTGGCGTGCATGAAGCGGTTCCCGGCCTCGTCGCCACCCACGCCATCCGCGTCGGCCAGCCACTCTCACTGCTCGC + AA11AC1>3@DC1F1111000A0/A///BB#############################################################################################################################################################################################################################This read has more consistent quality at its first than at the end but still has a range of quality scores, most of them are low. We will look at variations in position-based quality in just a moment.
In real life, you won’t be assessing the quality of your reads by visually inspecting your FASTQ files. Instead, you’ll use a software program to assess read quality and filter out poor reads. We’ll first use a program called FastQC to visualize the quality of our reads. Later in our workflow, we’ll use another program to filter out poor-quality reads.
First, let’s make available our metagenomics software:
Activating an environment
Environments are part of a bioinformatic tendency to do reproducible research; they are a way to share and maintain our programs in their needed versions used for a pipeline with our colleagues and our future self. FastQC has not been activated in the (base) environment, but this AWS instance came with an environment called metagenomics. We need to activate it in order to start using FastQC.
We will use Conda as our environment manager.
Conda is an open-source package and environment management system that runs on Windows,
macOS and Linux. Conda environments are activated with the conda activate direction:
$ conda activate metagenomics
After the environment has been activated, a label is shown before the $ sign.
(metagenomics) $
Now, if we call FastQC, a long help page will be displayed on our screen.
$ fastqc -h
FastQC - A high throughput sequence QC analysis tool
SYNOPSIS
fastqc seqfile1 seqfile2 .. seqfileN
fastqc [-o output dir] [--(no)extract] [-f fastq|bam|sam]
[-c contaminant file] seqfile1 .. seqfileN
DESCRIPTION
FastQC reads a set of sequence files and produces from each one a quality
control report consisting of many different modules, each one of
which will help to identify a different potential type of problem in your
data.
.
.
.
If FastQC is not installed, then you would expect to see an error like
The program 'fastqc' is currently not installed. You can install it by typing:
sudo apt-get install fastqc
If this happens, check with your instructor before trying to install it.
Assessing quality using FastQC
FastQC has several features that can give you a quick impression of any problems your data may have, so you can consider these issues before moving forward with your analyses. Rather than looking at quality scores for each read, FastQC looks at quality collectively across all reads within a sample. The image below shows one FastQC-generated plot that indicates a very high-quality sample:
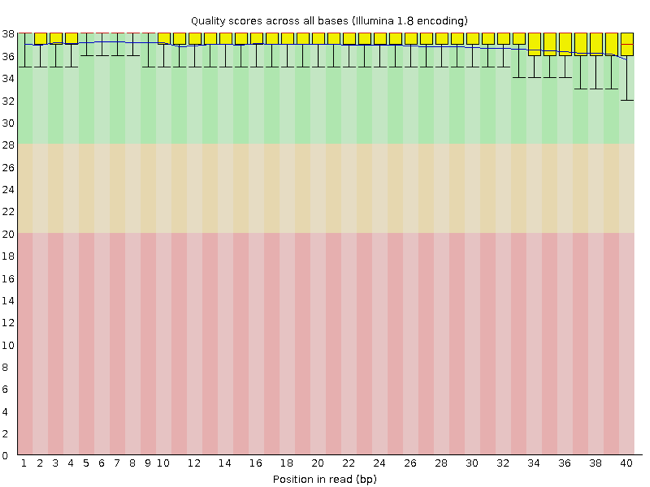
The x-axis displays the base position in the read, and the y-axis shows quality scores. In this example, the sample contains reads that are 40 bp long. This length is much shorter than the reads we are working on within our workflow. For each position, there is a box-and-whisker plot showing the distribution of quality scores for all reads at that position. The horizontal red line indicates the median quality score, and the yellow box shows the 1st to 3rd quartile range. This range means that 50% of reads have a quality score that falls within the range of the yellow box at that position. The whiskers show the whole range covering the lowest (0th quartile) to highest (4th quartile) values.
The quality values for each position in this sample do not drop much lower than 32, which is a high-quality score. The plot background is also color-coded to identify good (green), acceptable (yellow) and bad (red) quality scores.
Now let’s look at a quality plot on the other end of the spectrum.
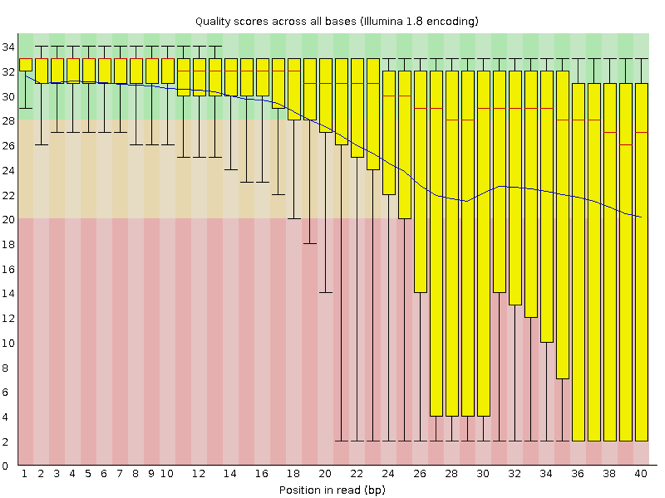
The FastQC tool produces several other diagnostic plots to assess sample quality and the one plotted above. Here, we see positions within the read in which the boxes span a much more comprehensive range. Also, quality scores drop pretty low into the “bad” range, particularly on the tail end of the reads.
Running FastQC
We will now assess the quality of the reads that we downloaded. First, make sure you’re still in the untrimmed_fastq directory.
$ cd ~/dc_workshop/data/untrimmed_fastq/
Exercise 2: Looking at metadata about the untrimmed-files
To know which files have more data, you need to see metadata about untrimmed files. In files, metadata includes owners of the file, state of the write, read, and execute permissions, size, and modification date. Using the
lscommand, how would you get the size of the files in theuntrimmed_fastq\directory?
(Hint: Look at the options for thelscommand to see how to show file sizes.)
a)ls -a
b)ls -S
c)ls -l
d)ls -lh
e)ls -ahlSSolution
a) No. The flag `-a` shows all the contents, including hidden files and directories, but not the sizes. b) No. The flag `-S` shows the content Sorted by size, starting with the most extensive file, but not the sizes. c) Yes. The flag `-l` shows the contents with metadata, including file size. Other metadata are permissions, owners, and modification dates. d) Yes. The flag `-lh` shows the content with metadata in a human-readable manner. e) Yes. The combination of all the flags shows all the contents with metadata, including hidden files, sorted by size.ls -ahls-rw-r--r-- 1 dcuser dcuser 24M Nov 26 21:34 JC1A_R1.fastq.gz -rw-r--r-- 1 dcuser dcuser 24M Nov 26 21:34 JC1A_R2.fastq.gz -rw-r--r-- 1 dcuser dcuser 616M Nov 26 21:34 JP4D_R1.fastq -rw-r--r-- 1 dcuser dcuser 203M Nov 26 21:35 JP4D_R2.fastq.gzFour FASTQ files oscillate between 24M (24MB) to 616M. The largest file is JP4D_R1.fastq with 616M.
FastQC can accept multiple file names as input, and on both zipped and unzipped files,
so we can use the \*.fastq*wildcard to run FastQC on all FASTQ files in this directory.
$ fastqc *.fastq*
You will see an automatically updating output message telling you the progress of the analysis. It will start like this:
Started analysis of JC1A_R1.fastq.gz
Approx 5% complete for JC1A_R1.fastq.gz
Approx 10% complete for JC1A_R1.fastq.gz
Approx 15% complete for JC1A_R1.fastq.gz
Approx 20% complete for JC1A_R1.fastq.gz
Approx 25% complete for JC1A_R1.fastq.gz
Approx 30% complete for JC1A_R1.fastq.gz
Approx 35% complete for JC1A_R1.fastq.gz
It should take around five minutes for FastQC to run on all four of our FASTQ files. When the analysis completes, your prompt will return. So your screen will look something like this:
Approx 80% complete for JP4D_R2.fastq.gz
Approx 85% complete for JP4D_R2.fastq.gz
Approx 90% complete for JP4D_R2.fastq.gz
Approx 95% complete for JP4D_R2.fastq.gz
Analysis complete for JP4D_R2.fastq.gz
$
The FastQC program has created several new files within our
data/untrimmed_fastq/ directory.
$ ls
JC1A_R1_fastqc.html JC1A_R2_fastqc.html JP4D_R1.fastq JP4D_R2_fastqc.html TruSeq3-PE.fa
JC1A_R1_fastqc.zip JC1A_R2_fastqc.zip JP4D_R1_fastqc.html JP4D_R2_fastqc.zip
JC1A_R1.fastq.gz JC1A_R2.fastq.gz JP4D_R1_fastqc.zip JP4D_R2.fastq.gz
For each input FASTQ file, FastQC has created a .zip file and a
.html file. The .zip file extension indicates that this is
a compressed set of multiple output files. We’ll be working
with these output files soon. The .html file is a stable webpage
displaying the summary report for each of our samples.
We want to keep our data files and our results files separate, so we
will move these
output files into a new directory within our results/ directory.
$ mkdir -p ~/dc_workshop/results/fastqc_untrimmed_reads
$ mv *.zip ~/dc_workshop/results/fastqc_untrimmed_reads/
$ mv *.html ~/dc_workshop/results/fastqc_untrimmed_reads/
Now we can navigate into this results directory and do some closer inspection of our output files.
$ cd ~/dc_workshop/results/fastqc_untrimmed_reads/
Viewing the FastQC results
If we were working on our local computers, we’d be able to look at each of these HTML files by opening them in a web browser. However, these files are currently sitting on our remote AWS instance, where our local computer can’t see them. If we are working with the terminal provided by r-studio we can either: select the html files and with the secondary click chose the option open in a browser or export the files to our local computer as we learned in the Introduction to the Command Line lesson.
Exercise 3: Downloading files by scp (optional)
Suppose you only have access to a terminal and there is not a web browser available at the AWS remote machine. The following are the steps needed to download your files to your computer. Observe the third step and answer false/true for each question about the
scpcommand.
- Open a new terminal on your local computer.
- Make a new local directory on our computer to store the HTML files
$ mkdir -p ~/Desktop/fastqc_html- Transfer our HTML files to our local computer using
scp.$ scp dcuser@ec2-34-238-162-94.compute-1.amazonaws.com:~/dc_workshop/results/fastqc_untrimmed_reads/*.html ~/Desktop/fastqc_html
A)dcuseris your local user
B)ec2-34-238-162-94.compute-1.amazonaws.comis the address of your remote machine
C) the current adress of the file goes after the second space in thescpcommand.
D)~/dc_workshop/results/fastqc_untrimmed_reads/*.htmlis the path of the file you want to download
E)~/Desktop/fastqc_htmlis a remote path.
F):Divides the host name of your local computer and the path of the file.Solution
A) False.
dcuseris your remote user.
B) True.ec2-34-238-162-94.compute-1.amazonaws.comis the adress of your remote machine
C) False. The current adress of the file goes after the first space in thescpcommand.
D) True.~/dc_workshop/results/fastqc_untrimmed_reads/*.htmlis the path of the file you want to download in the remote machine. E) False.~/Desktop/fastqc_htmlis a local path where your file will be downloaded. F) False.:Divides the host name of a remote computer and the path of the file on the remote computer.You should see a status output like this:
JC1A_R1_fastqc.html 100% 253KB 320.0KB/s 00:00 JC1A_R2_fastqc.html 100% 262KB 390.1KB/s 00:00 JP4D_R1_fastqc.html 100% 237KB 360.8KB/s 00:00 JP4D_R2_fastqc.html 100% 244KB 385.2KB/s 00:00
Now we can open the 4 HTML files.
Depending on your system, you should be able to select and open them all at once via a right-click menu in your file browser.
Exercise 4: Discuss the quality of sequencing files
Discuss your results with a neighbor. Which sample(s) looks the best per base sequence quality? Which sample(s) look the worst?
Solution
All of the reads contain usable data, but the quality decreases toward the end of the reads. File JC1A_R2_fastqc shows the lowest quality.
Decoding the other FastQC outputs
We’ve now looked at quite a few “Per base sequence quality” FastQC graphs, but there are nine other graphs that we haven’t talked about! Below we have provided a brief overview of interpretations for each plot. For more information, please see the FastQC documentation here
- Per tile sequence quality: the machines that perform sequencing are divided into tiles. This plot displays patterns in base quality along these tiles. Consistently low scores are often found around the edges, but hot spots could also occur in the middle if an air bubble was introduced during the run.
- Per sequence quality scores: a density plot of quality for all reads at all positions. This plot shows what quality scores are most common.
- Per base sequence content: plots the proportion of each base position over all of the reads. Typically, we expect to see each base roughly 25% of the time at each position, but this often fails at the beginning or end of the read due to quality or adapter content.
- Per sequence GC content: a density plot of average GC content in each of the reads.
- Per base N content: the percent of times that ‘N’ occurs at a position in all reads. If there is an increase at a particular position, this might indicate that something went wrong during sequencing.
- Sequence Length Distribution: the distribution of sequence lengths of all reads in the file. If the data is raw, there is often a sharp peak; however, if the reads have been trimmed, there may be a distribution of shorter lengths.
- Sequence Duplication Levels: a distribution of duplicated sequences. In sequencing, we expect most reads to only occur once. If some sequences are occurring more than once, it might indicate enrichment bias (e.g. from PCR). This might not be true if the samples are high coverage (or RNA-seq or amplicon).
- Overrepresented sequences: a list of sequences that occur more frequently than would be expected by chance.
- Adapter Content: a graph indicating where adapter sequences occur in the reads.
- K-mer Content: a graph showing any sequences which may show a positional bias within the reads.
Working with the FastQC text output
Now that we’ve looked at our HTML reports getting a feel for the data,
let’s look more closely at the other output files. Go back to the tab
in your terminal program that is connected to your AWS instance
(the tab label will start with dcuser@ip) and make sure you’re in
our results subdirectory.
$ cd ~/dc_workshop/results/fastqc_untrimmed_reads/
$ ls
JC1A_R1_fastqc.html JP4D_R1_fastqc.html
JC1A_R1_fastqc.zip JP4D_R1_fastqc.zip
JC1A_R2_fastqc.html JP4D_R2_fastqc.html
JC1A_R2_fastqc.zip JP4D_R2_fastqc.zip
Our .zip files are compressed files. Each contains multiple
different types of output files for a single input FASTQ file. To
view the contents of a .zip file, we can use the program unzip
to decompress these files. Let’s try doing them all at once using a
wildcard.
$ unzip *.zip
Archive: JC1A_R1_fastqc.zip
caution: filename not matched: JC1A_R2_fastqc.zip
caution: filename not matched: JP4D_R1_fastqc.zip
caution: filename not matched: JP4D_R2_fastqc.zip
This decompression didn’t work. It identified the first file and got a warning
message for the other .zip files. This is because unzip
expects to get only one zip file as input. We could go through and
unzip each file one at a time, but this is very time-consuming and
error-prone. Someday you may have 500 files to unzip!
A more efficient way is to use a for loop like we learned in the Command Line lesson to iterate through all of
our .zip files. Let’s see what that looks like, and then we’ll
discuss what we’re doing with each line of our loop.
$ for filename in *.zip
> do
> unzip $filename
> done
In this example, the input is the four filenames (one filename for each of our .zip files).
Each time the loop iterates, it will assign a file name to the variable filename
and run the unzip command.
The first time through the loop,
$filename is JC1A_R1_fastqc.zip.
The interpreter runs the command unzip on JC1A_R1_fastqc.zip.
For the second iteration, $filename becomes
JC1A_R2_fastqc.zip. This time, the shell runs unzip on JC1A_R2_fastqc.zip.
It then repeats this process for the other .zip files in our directory.
When we run the for loop, you will see an output that starts like this:
Archive: JC1A_R1_fastqc.zip
creating: JC1A_R1_fastqc/
creating: JC1A_R1_fastqc/Icons/
creating: JC1A_R1_fastqc/Images/
inflating: JC1A_R1_fastqc/Icons/fastqc_icon.png
inflating: JC1A_R1_fastqc/Icons/warning.png
inflating: JC1A_R1_fastqc/Icons/error.png
inflating: JC1A_R1_fastqc/Icons/tick.png
inflating: JC1A_R1_fastqc/summary.txt
inflating: JC1A_R1_fastqc/Images/per_base_quality.png
inflating: JC1A_R1_fastqc/Images/per_tile_quality.png
inflating: JC1A_R1_fastqc/Images/per_sequence_quality.png
inflating: JC1A_R1_fastqc/Images/per_base_sequence_content.png
inflating: JC1A_R1_fastqc/Images/per_sequence_gc_content.png
inflating: JC1A_R1_fastqc/Images/per_base_n_content.png
inflating: JC1A_R1_fastqc/Images/sequence_length_distribution.png
inflating: JC1A_R1_fastqc/Images/duplication_levels.png
inflating: JC1A_R1_fastqc/Images/adapter_content.png
inflating: JC1A_R1_fastqc/fastqc_report.html
inflating: JC1A_R1_fastqc/fastqc_data.txt
inflating: JC1A_R1_fastqc/fastqc.fo
The unzip program is decompressing the .zip files and creates
a new directory (with subdirectories) for each of our samples, to
store all of the different output that is produced by FastQC. There
are a lot of files here. We’re going to focus on the
summary.txt file.
If you list the files in our directory, now you will see the following:
$ ls
JC1A_R1_fastqc JP4D_R1_fastqc
JC1A_R1_fastqc.html JP4D_R1_fastqc.html
JC1A_R1_fastqc.zip JP4D_R1_fastqc.zip
JC1A_R2_fastqc JP4D_R2_fastqc
JC1A_R2_fastqc.html JP4D_R2_fastqc.html
JC1A_R2_fastqc.zip JP4D_R2_fastqc.zip
The .html files and the uncompressed .zip files are still present,
but now we also have a new directory for each sample. We can
see that it’s a directory if we use the -F flag for ls.
$ ls -F
JC1A_R1_fastqc/ JP4D_R1_fastqc/
JC1A_R1_fastqc.html JP4D_R1_fastqc.html
JC1A_R1_fastqc.zip JP4D_R1_fastqc.zip
JC1A_R2_fastqc/ JP4D_R2_fastqc/
JC1A_R2_fastqc.html JP4D_R2_fastqc.html
JC1A_R2_fastqc.zip JP4D_R2_fastqc.zip
Let’s see what files are present within one of these output directories.
$ ls -F JC1A_R1_fastqc/
fastqc_data.txt fastqc.fo fastqc_report.html Icons/ Images/ summary.txt
Use less to preview the summary.txt file for this sample.
$ less JC1A_R1_fastqc/summary.txt
PASS Basic Statistics JC1A_R1.fastq.gz
FAIL Per base sequence quality JC1A_R1.fastq.gz
PASS Per tile sequence quality JC1A_R1.fastq.gz
PASS Per sequence quality scores JC1A_R1.fastq.gz
WARN Per base sequence content JC1A_R1.fastq.gz
FAIL Per sequence GC content JC1A_R1.fastq.gz
PASS Per base N content JC1A_R1.fastq.gz
PASS Sequence Length Distribution JC1A_R1.fastq.gz
FAIL Sequence Duplication Levels JC1A_R1.fastq.gz
PASS Overrepresented sequences JC1A_R1.fastq.gz
FAIL Adapter Content JC1A_R1.fastq.gz
The summary file gives us a list of tests that FastQC ran and tells
us whether this sample passed, failed, or is borderline (WARN). Remember, to quit from less, you must type q.
Documenting our work
We can make a record of the results we obtained for all our samples
by concatenating all of our summary.txt files into a single file
using the cat command. We’ll call this fastqc_summaries.txt and store
it to ~/dc_workshop/docs.
$ mkdir -p ~/dc_workshop/docs
$ cat */summary.txt > ~/dc_workshop/docs/fastqc_summaries.txt
Exercise 4: Quality tests
Which samples failed at least one of FastQC’s quality tests? What test(s) did those samples failed
Solution
We can get the list of all failed tests using
grep.$ cd ~/dc_workshop/docs $ grep FAIL fastqc_summaries.txtFAIL Per base sequence quality JC1A_R1.fastq.gz FAIL Per sequence GC content JC1A_R1.fastq.gz FAIL Sequence Duplication Levels JC1A_R1.fastq.gz FAIL Adapter Content JC1A_R1.fastq.gz FAIL Per base sequence quality JC1A_R2.fastq.gz FAIL Per sequence GC content JC1A_R2.fastq.gz FAIL Sequence Duplication Levels JC1A_R2.fastq.gz FAIL Adapter Content JC1A_R2.fastq.gz FAIL Per base sequence content JP4D_R1.fastq FAIL Adapter Content JP4D_R1.fastq FAIL Per base sequence quality JP4D_R2.fastq.gz FAIL Per base sequence content JP4D_R2.fastq.gz FAIL Adapter Content JP4D_R2.fastq.gz
Quality of large datasets
Explore MultiQC if you want a tool that can show the quality of many samples at once.
Quality Encodings Vary
Although we’ve used a particular quality encoding system to demonstrate the interpretation of read quality, different sequencing machines use different encoding systems. This means that depending on which sequencer you use to generate your data, a
#may not indicate a poor quality base call.This mainly relates to older Solexa/Illumina data. However, it’s essential that you know which sequencing platform was used to generate your data to tell your quality control program which encoding to use. If you choose the wrong encoding, you run the risk of throwing away good reads or (even worse) not throwing away bad reads!
Bonus Exercise: Automating a quality control workflow
If you lose your FastQC analysis results. How would you do it again but faster than the first time? As we have seen in a previous lesson, making scripts for repetitive tasks is a very efficient practice during bioinformatic pipelines.
Solution
Make a new script with nano
nano quality_control.shPaste inside the commands that we used along with
echocommands that shows you how the script is running.set -e # This will ensure that our script will exit if an error occurs cd ~/dc_workshop/data/untrimmed_fastq/ echo "Running FastQC ..." fastqc *.fastq* mkdir -p ~/dc_workshop/results/fastqc_untrimmed_reads echo "Saving FastQC results..." mv *.zip ~/dc_workshop/results/fastqc_untrimmed_reads/ mv *.html ~/dc_workshop/results/fastqc_untrimmed_reads/ cd ~/dc_workshop/results/fastqc_untrimmed_reads/ echo "Unzipping..." for filename in *.zip do unzip $filename done echo "Saving summary..." mkdir -p ~/dc_workshop/docs cat */summary.txt > ~/dc_workshop/docs/fastqc_summaries.txtIf we were to run this script, it would ask us for confirmation to redo several steps because we already did all of them. If you want to, you can run it to check that it works, but it is not necessary if you already completed every step of the previous episode.
Key Points
It is important to know the quality of our data to make decisions in the subsequent steps.
FastQC is a program that allows us to know the quality of FASTQ files.
forloops let you perform the same operations on multiple files with a single command.