Genomas a partir de metagenomas
La metagenómica hace referencia al estudio de todo el ADN de los organismos que se encuentran en un ambiente. La secuenciación de este material genético produce lecturas que pueden ensamblarse para conocer la diversidad microbiana y sus funciones.
Típicamente los metagenomas pueden estudiarse mediante dos aproximaciones:
- La clasificación taxonómica de contigs o lecturas y la inferencia metabólica de los contigs.
- La reconstrucción de genomas a a partir de metagenomas (MAGs), clasificación taxonómica y la inferencia metabólica de los MAGs.
En este apartado nos enfocaremos en la segunda aproximación. Los MAGs se reconstruyen a partir de un ensamble metagenómico,
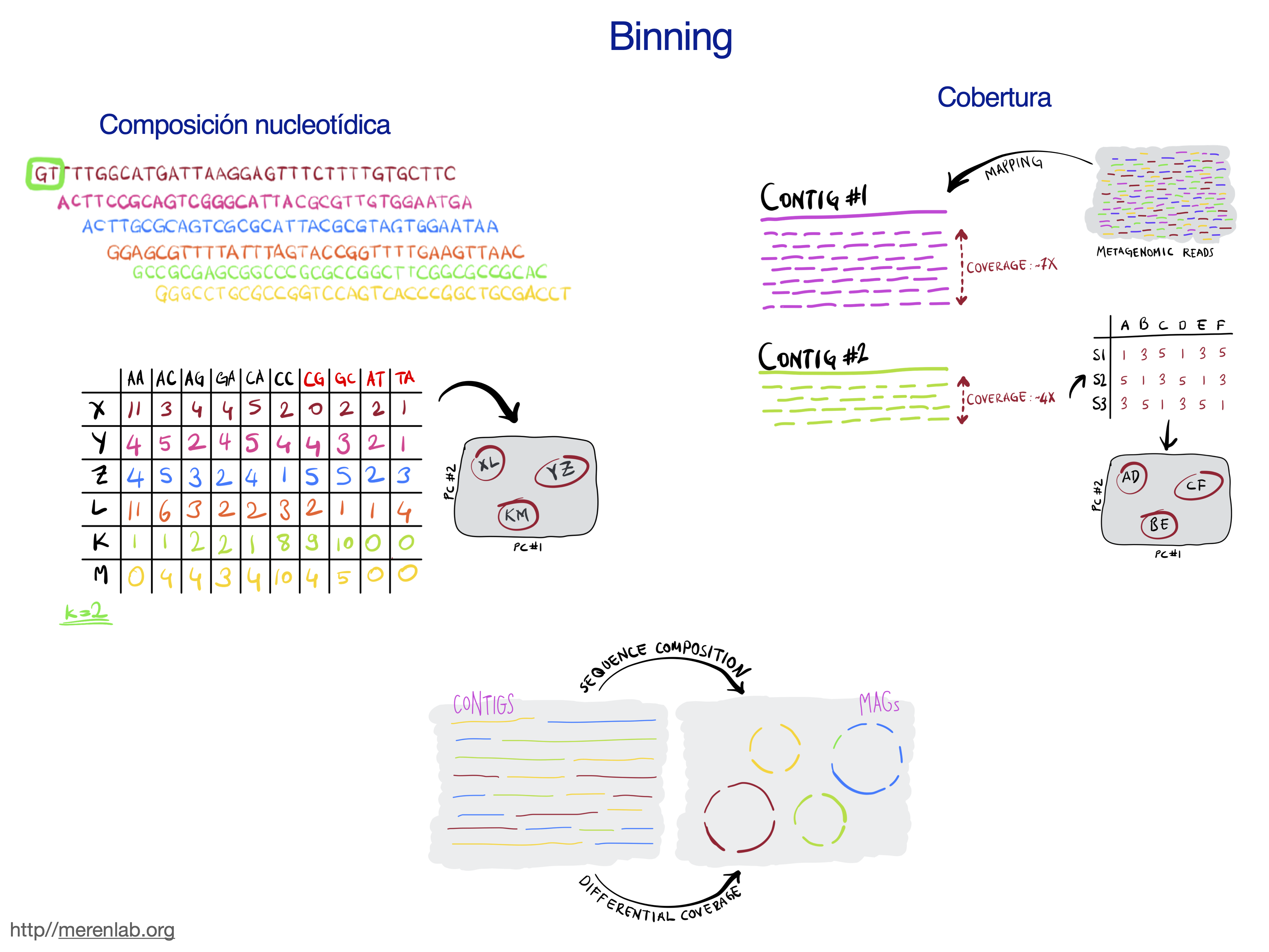
los contigs de dicho ensamble se agrupan mediante la información de cobertura y frecuencia de tetranucleótidos.
Esta agrupación puede generar errores, por lo que es indispensable evaluar la calidad de los MAGs mediante la completitud
y redundancia de genes de copia única MerenLab y col.
Para obtener MAGs podemos seguir el siguiente flujo de análisis:
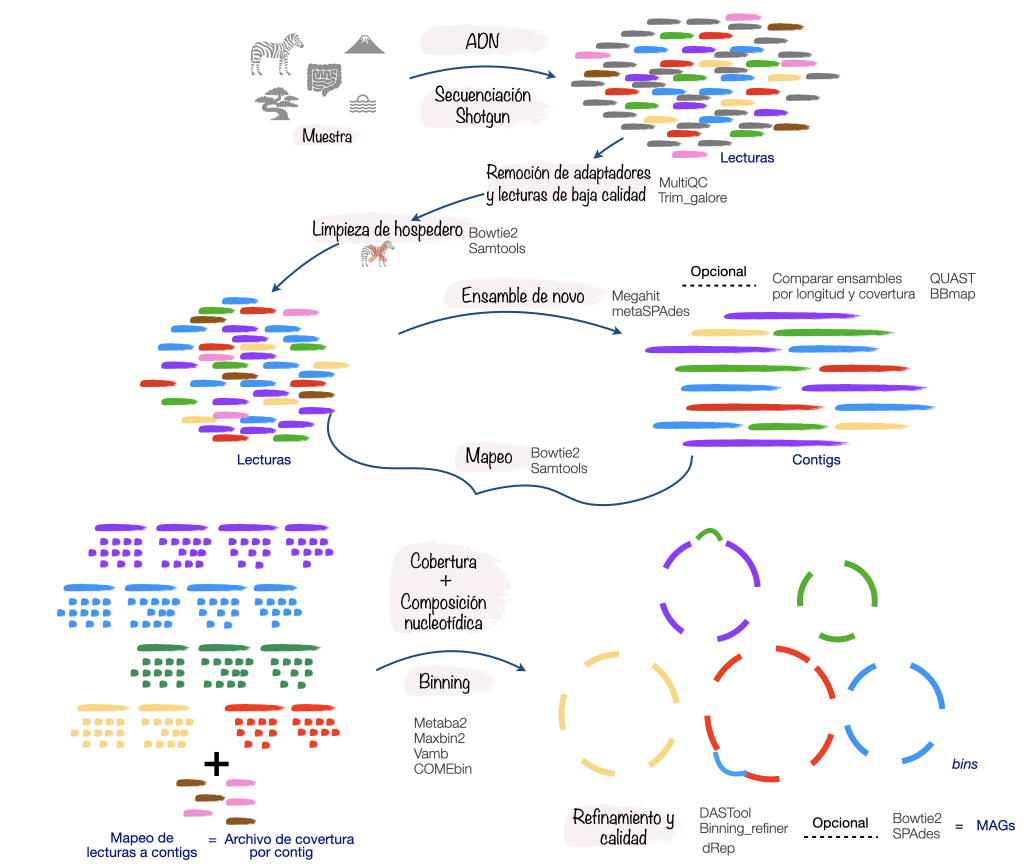
Ya que discutimos como seguir un flujo de análisis para reconstruir genomas entremos en acción, para ello analizaremos el metagenoma del pozol.
El pozol
El pozol es un alimento ácido, fermentado a partir de maíz nixtamalizado, de importancia económica y cultural, se consume desde tiempos prehispánicos y se ha estudiado desde los años 50s.

Algunos puntos importantes que conocemos son:
* Es muy nutritivo, tiene un alto contenido de aminoácidos esenciales.
* Es considerado como prebiótico, contiene fibras solubles y microorganismos benéficos para la salud intestinal humana.
Resolvamos preguntas biológicas mediante Metagenómica centrada en genomas
🧬🔊🦠 Imaginemos que se quiere impulsar la producción de esta bebida y para ello necesitan saber todo acerca de su naturaleza microbiana.
Una importante industria alimenticia los contacta como expertos en ecología microbiana y les pide ayuda para descubrir los siguientes puntos:
* ¿Cómo ocurre la bioconversión del maíz durante la fermentación, quién participa y cómo lo hace? ¿Qué funciones metabólicas están ocurriendo?
* ¿Cambia la comunidad microbiana a lo largo del proceso?
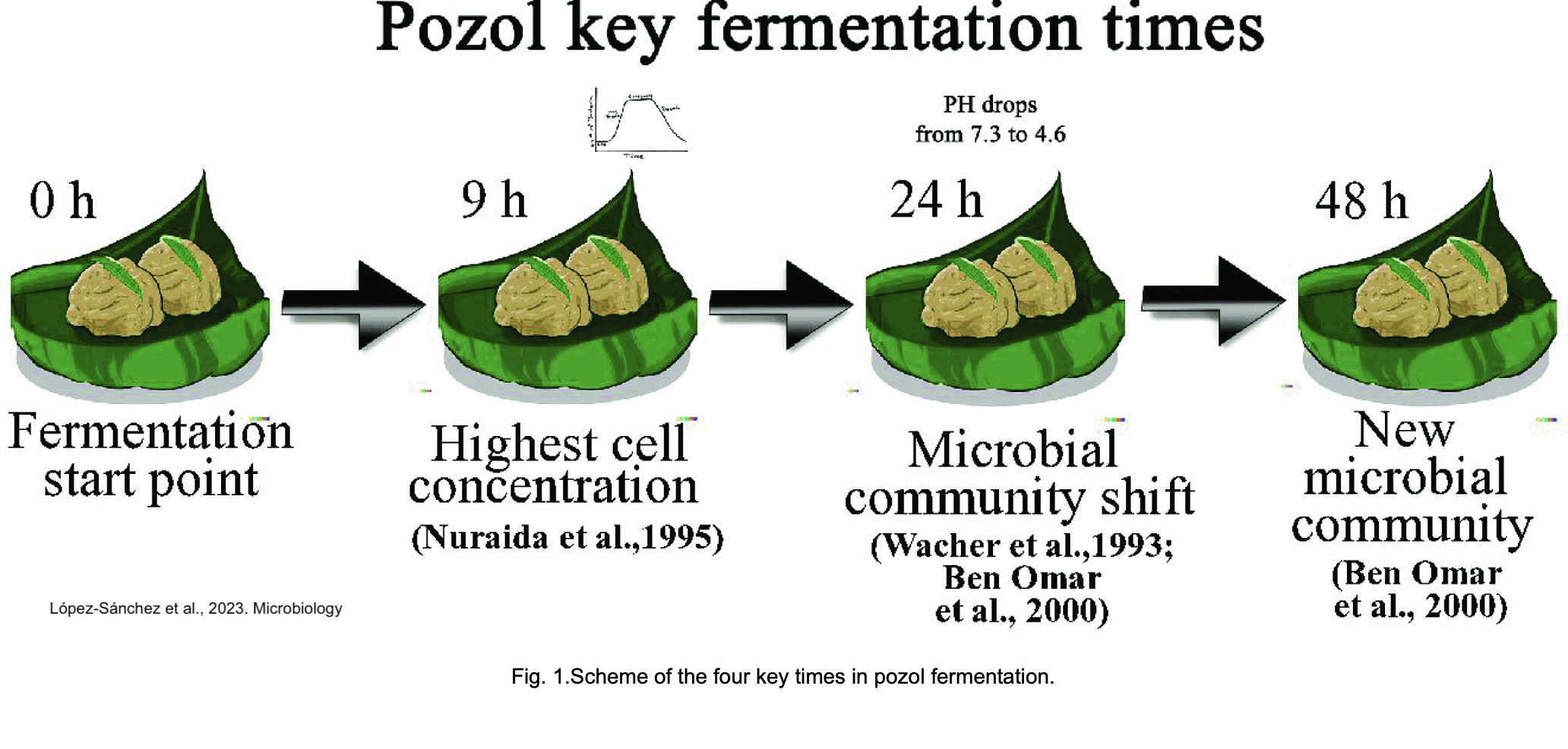
La empresa secuenció cuatro puntos de fermentación de muestras que se obtuvieron en un mercado de Tabasco. Las muestras se secuenciaron con Illumina NextSeq500 con lecturas pareadas de 75 pb. Los datos están públicos bajo el Bioproject: PRJNA648868
Limpieza de hospedero
Como las muestras contienen maíz, es indispensable remover las lecturas que correspondan a su genoma, no hacerlo producirá un ensamble muy fragmentado, mayoritariamente del maíz y poco microbiano. El autor del artículo amablemente nos proporcionó sus muestras libres del maíz y el código que usó para ello está disponible en un repositorio público de GitHub.
El artículo: López-Sánchez et al., 2023. Analysing the dynamics of the bacterial community in pozol, a Mexican fermented corn dough.
Espacio de trabajo
- Entra a tu cuenta en el servidor y sitúate en tu
$HOME - Obten los datos y la estructura de tu directorio del proyecto
- Entra al directorio del proyecto
# ve al $HOME
cd
# descarga
#wget https://zenodo.org/records/13911654/files/taller_metagenomica_pozol.tar.gz?download=1 -O taller_metagenomica_pozol.tar.gz
# descomprime
#tar -xvzf taller_metagenomica_pozol.tar.gz
# Entra al directorio del proyecto
cd taller_metagenomica_pozol
Si en algún momento te pierdes entre directorios, puedes regresar al espacio principal asi:
Directorio principal del proyecto
cd && cd taller_metagenomica_pozol/
Cómo vamos a trabajar durante el taller?
Reglas del juego
- En este tutorial haremos el ejemplo corriendo la muestra de 48 hrs.
- Se formaran 6 equipos (2 de los tiempos 0, 9 y 24 hrs).
- Los equipos discutirán y presentarán sus resultados cuando se indique en el tutorial.
La presente práctica sólo es una representación del flujo de trabajo para el análisis metagenómico, sin embargo, `no sustituye los manuales` de cada programa y el flujo puede variar dependiendo del tipo de datos y pregunta de investigación. De hecho para fines del taller, con frecuencia se utilizan las lineas de comando más simples para eficientar tiempo y recursos, tómalo en cuenta.
Cada programa tiene una ayuda y un manual de usuario, es importante revisarlo y conocer cada parámetro que se ejecute. En terminal se puede consultar el manual con el comando man y también se puede consultar la ayuda con -h o --help, por ejemplo fastqc -h.
🧠 Para tenerlo presente
En bioinformática cualquier línea de comandos generará un resultado, de ahí a que esos resultados sean correctos puede haber una gran diferencia. En cada paso detente a revisar la información de cada programa, lee el manual, visita foros de ayuda y selecciona los argumentos que se ajusten a las necesidades de tus datos.